Type of Group 1 PAH
Number of patients (% female)
Ratio of females:males
All patients with Group 1 PAH
2525 (79.5 %)
3.9:1
IPAH
1166 (80.3 %)
4.1:1
Drugs/Toxins-PAH
134 (84.3 %)
5.4:1
CHD-PAH
250 (73.6 %)
2.8:1
CTD-PAH
639 (90.1)
9.1:1
In the early 2000s, French investigators enrolled 674 cases of PAH from 17 university hospitals across France over a 12-month period. They found a higher proportion of females among prevalent cases (67.1 % of prevalent cases) [6]. But, among new diagnoses (incident cases; n = 121) of PAH, the percentage of females was lower, at 57.0 % within that cohort (n = 121). While not a dramatic difference, the discrepancy in female predominance between incident and prevalent cases does suggest a survival benefit to being female over male consistent with the REVEAL data. Furthermore, in a follow-up study in France, those same investigators showed that heritable PAH (HPAH), idiopathic PAH (IPAH), and anorexigen-associated PAH all had improved survival for females compared to males [8, 9].
While the differences in survival according to sex remain unclear, it is known that survival in PAH is strongly influenced by the response and performance of the right ventricle to load stress [21]. While sex and sex hormone levels have been extensively studied in left heart failure, the role of sex hormones in right ventricular (RV) function irrespective of disease status is relatively understudied. Recently, some clues have emerged from the work of investigators of the Multi-Ethnic Study of Atherosclerosis (MESA) program. MESA is a multicenter prospective cohort study designed to investigate subclinical cardiovascular disease in White, African-American, Hispanic, and Chinese subjects [22]. While the MESA study is not specific to PAH (and in fact is a study of reportedly healthy subjects), its findings in postmenopausal women suggested that higher levels of circulating estradiol (one of the main “parent compound” estrogens) are associated with higher RV systolic function . In both healthy postmenopausal women and men, levels of androgens (testosterone and dehydroepiandrosterone) are associated with higher RV mass and larger RV volumes [23]. Among PAH patients, females had a higher RV ejection fraction at the time of diagnosis than male patients [24]. Thus, there is growing data to suggest that female sex may negatively influence pulmonary vascular disease pathogenesis, but positively influence right heart adaptation to the stress of pulmonary hypertension.
The Association Between Sex Hormones and Human PAH
The long-standing understanding that PAH is a disease influenced by sex suggests that factors related to genetic differences between the sexes (e.g., chromosomal differences, XX and XY) and/or hormonal variations contribute to disease pathogenesis and perhaps progression. There is a growing body of investigation exploring these possibilities.
Genetic Differences Related to the Sex Chromosomes
To date, few studies have investigated sex chromosomal differences in humans . The X and Y chromosomes are vital contributors of genetic data to the human and other genomes. The X chromosome contributes about 155 million base pairs of DNA, representing approximately 5 % of the total DNA in cells. The Y chromosome contributes about 59 million base pairs of DNA , representing approximately 2 % of the total DNA in cells. Most genome-wide association studies have underinvestigated the contribution of sex chromosomes in all disease states due to the challenge of integrating the sex chromosomes using bioinformatics . There is a line of investigation suggestive that the presence of the Y chromosome is protective in hypoxia-associated mice models of PH, suggesting that genetic factors on the Y chromosome are relevant (ATS 2015 Conference: Am J Respir Crit Care Med 191;2015:A4100), which is an intriguing hypothesis.
Differences with Regard to Sex Hormones
Unlike the sex chromosomes, which hopefully will prompt increased interest over time, there has been tremendous research interest in the sex hormones in the past decade. Traditionally, female sex hormones, in particular estrogens , have been perceived as protective of pulmonary vascular reactivity, inflammation, and remodeling. This has largely derived from animal model work on the disease over the past 30 years. In humans, it is notable that even for diseases with a global female predominance irrespective of PAH (e.g., connective tissue disease), the predominance of females among the subset of subjects with PAH is substantial. For example, the REVEAL study found that among PAH patients with connective tissue disease, nine females were represented for every one male [20]. Interestingly, unlike other forms of PAH, connective tissue disease-associated PAH is more often associated with older diagnosis, in particular postmenopausal women [20, 25]. The relationship between this finding and menopausal status could suggest that hormones are protective in connective tissue disease patients. In support of the hypothesis that female sex hormones are protective, a retrospective study of 61 scleroderma patients without echocardiographic evidence of pulmonary hypertension at the time of menopause demonstrated the development of PAH in the subsequent postmenopausal years among 20 patients was 0 % (mean duration of follow-up was 7.2 ± 3.5 years) for those using hormone replacement therapy; in contrast, 8 out of 41 patients (19.5 %) developed PAH after a similar period of time postmenopause (7.5 ± 3.9 years) but without any hormone replacement therapy [26]. This single study does suggest an association between hormone replacement therapy and protection among those with connective tissue disease, but certainly the topic requires further study.
There is more data to suggest that sex hormones, in particular estrogens, contribute in a detrimental manner to PAH. First, idiopathic and heritable PAH forms, as well as anorexigen-associated, are clinically known to manifest during or soon after pregnancy, and pharmacologic exposure to estrogens has been described in PAH patients although not systematically studied to date [27–30]. Most PAH clinicians caution patients to avoid pregnancy, in part due to the significant changes to blood volume and circulation. In addition, while the ramifications are unknown, there are significant increases in estrogens and progestins during pregnancy related to placental production [2]. Among female patients with portopulmonary PAH, genetic variations in estrogen signaling, as well as enhanced plasma estradiol levels, are associated with PAH [31]. Similarly, genetic variations in estrogen metabolism, as well as estrogen metabolite levels, are associated with heritable PAH in females due to mutations in bone morphogenetic protein receptor type 2 (BMPR2) ; recently, males with BMPR2 gene mutations had similar irregularities in estrogen metabolites [32, 33].
From a broad epidemiologic perspective, there is a paucity of data concerning sex hormone exposures and PAH. However, a recent epidemiologic study sought to address this deficiency, using 88 patients attending the Eighth International Pulmonary Hypertension Association Conference. While lacking a control group for comparison, this study used sex hormone exposure questionnaires and found that 81 % of women with PAH reported prior use of any hormone therapy, with 70 % reporting exogenous hormone use for greater than 10 years . This supported an association of PAH with estrogen-containing compounds [34].
The Pulmonary Vasculature : Effects of Sex Hormones
Estrogens, progestins, and androgens have multiple effects on the pulmonary vasculature. Overall, it is difficult to label any one sex hormone as “detrimental” or “protective.” On the contrary, the cell type, situation, and duration are likely highly important but still explain only a proportion of the variability. While this may not be the case for all forms of PH, for the PAH condition estrogens may be acutely protective but detrimental on a chronic basis. The data concerning other sex hormones is even more insufficiently elucidated.
Acutely, estrogens are likely protective for the pulmonary vasculature. Smooth muscle cell activation by estradiol results in vasodilation and attenuation of the vasoconstrictor response caused by various stimuli, including hypoxia . In the physiologic state, variations in estrogen levels (for example, as occurs in menstruating females) are associated with the degree of pulmonary artery vasoconstriction in response to hypoxia—hypoxic vasoconstriction is reduced with exposure to increased circulating estrogen levels [35]. While the precise mechanism may be multifactorial, estradiol appears to stimulate the release of compounds known to promote pulmonary artery relaxation, including prostacyclin and nitric oxide (NO) acutely [11].
While estrogens exert their effects by a multitude of manners, the canonical pathways of estradiol-induced vasorelaxation occur via binding to one of two estrogen receptors: ERα and ERβ [36, 37]. This has been shown largely via animal model work, such as in the Sprague–Dawley rat model . In this system, the acute vasodilatory effects of estradiol occur via acute ligand binding by estradiol to both estrogen receptor α (ERα) and estrogen receptor β (ERβ). In this model system, direct binding of both ERα and ERβ mediated arterial vasorelaxation [38]. But in addition to ERα and ERβ , growing data suggest that estrogens can both directly bind to DNA to influence transcription and non-genomically signal via the g-coupled protein receptor GPR30. GPR30-mediated signaling promotes vascular relaxation [39].
Despite the evidence that estrogens, or at least estradiol, acutely relax the pulmonary vasculature, there is concern that on a chronic basis they may be detrimental. In particular, estradiol promotes inappropriate pulmonary vascular proliferation and cell damage on a chronic basis in vitro. Much of this work has evolved from the sex hormone cancer literature, although there is growing evidence using lung vascular cells . For example, estradiol is a potent mitogen which promotes proliferation of pulmonary vascular smooth muscle cells and other cell types; estrogen antagonism by the drug tamoxifen blocks this effect [40]. In addition, estradiol may directly modify angiogenesis in a number of ways, including the alteration of endothelial cell migration and function via GPR30. Angiogenesis characterizes severe PAH in humans and this could be a feature accentuated by estrogens.
While much of the focus of sex hormones in PAH has centered upon estrogens, androgens likely modify pulmonary vascular cell function and growth, as well. Both testosterone and dehydroepiandrosterone (DHEA) appear protective via induction of vasodilatation in studies of pulmonary vascular smooth muscle cells via acute antagonism of calcium signaling with augmentation of potassium channel-mediated relaxation in male and female cells [41, 42]. In the setting of hypoxia, DHEA protects human pulmonary artery smooth muscle cells via reduction of hypoxia-inducible factor 1α, which is a vasoconstrictive agent in the pulmonary vasculature, suggesting a protective influence. Intriguingly, DHEA inhibits Src/STAT3 activation in the pulmonary artery smooth muscle cells of patients with PAH; while results of this activation are incompletely elucidated, upregulation of both BMPR2 and miR-204 expression occurs and is relevant to pulmonary vascular homeostasis [43]. Overall, however, animal model data concerning the influence of androgens on the pulmonary hypertensive phenotype are mixed; for example, while male rodents are often more susceptible to most forms of experimental pulmonary hypertension, treatment with DHEA may be protective. As a result, a firm conclusion regarding the influence of androgens is difficult to provide at this time, pending additional animal studies which are currently under way [44].
Beyond Parent Compound Sex Hormones: Metabolites
While parent compound sex hormones likely play a role in pulmonary vascular homeostasis in health and disease, their metabolites likely contribute as well. The biology around the role of metabolites is even more incomplete, but of growing interest. Sex hormone metabolites likely mediate estrogenic and antiestrogenic effects on the pulmonary vasculature. In fact, inherent and iatrogenic variations in metabolism may account for the apparent contradictory influences of estrogens noted above. For example, while it appears that most parent compound estrogens (e.g., estradiol) are pro-proliferative on a chronic basis, not all estrogen metabolites share this impact. Depending on the estrogen metabolite , one can see disparate effects, and there may be variations according to cell type as with estradiol. The specifics of these differences have yet to be fully elucidated for parent compound sex hormones or their metabolites.
While androgens can be converted to estrogens, the metabolism of sex hormones is a complex process, largely mediated via the cytochrome P450 (CYP) family of enzymes both systemically and at a local level within the pulmonary vasculature (Fig. 5.1) [45, 46]. CYP1B1, for example, which oxidizes estradiol and estrone, is highly expressed in the lung and other organs [46]. Oxidation of estrogens also occurs by hydroxylation at the C-2, C-4, and C-16 position by CYP450 enzymes, followed by additional enzymatic reactions [47, 48]. It appears that “2-estrogens” are antimitogenic, while “16-estrogens ” stimulate cellular proliferation by constitutively activating the estrogen receptors and this has been demonstrated in the pulmonary vasculature [46]. “16-estrogens” may also damage the genome directly via the formation of unstable DNA adducts [49, 50]. As a result, individuals who produce a higher ratio of “16-estrogens” compared to other estrogen metabolites may be at increased risk of diseases that result from both the mitogenic and genotoxic effects of estrogens [51–56].
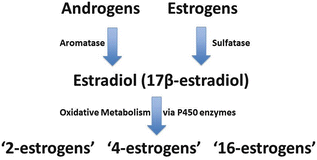
Fig. 5.1
Simplified schematic of sex hormones and estrogen metabolism. Sulfated estrogens, and androgens, are converted to parent compound estrogens including estradiol. Estradiol is metabolized by CYP450 enzyme activity into oxidized products, including those oxidized at the 2-, 4-, and 16-positions
Interest in the role of estrogens in PAH was redirected in the early 2000s due to the discovery by West et al. that CYP1B1 expression associates with PAH among BMPR2 mutation carriers. In fact, quantitative measures of CYP1B1 expression showed tenfold lower expression levels in female patients compared to healthy BMPR2 mutation carriers [57]. White and colleagues subsequently found enhanced CYP1B1 expression in the lungs of PAH patients without mutation [46].
As noted above, it was subsequently demonstrated that a specific urinary profile of estrogen metabolites is associated with PAH among female BMPR2 mutation carriers. Specifically, PAH patients had significantly lower ratio of 2-hydroxyestrogens (2-OHE1/2): 16-α-hydroxyestrone (16-αOHE1) compared to unaffected BMPR2 mutation carriers [32]. In addition, certain functional polymorphisms in CYP1B1 (a cytochrome P450 enzyme critical to estrogen metabolism) consistent with the urinary data are also associated with PAH in those subjects [32].
The influence of estrogen metabolites to PAH remains an area of active investigation. We and others believe that variations in estrogen metabolites influence the development of PAH , especially among those with an underlying disease risk. For example, estradiol and 16-αOHE1 both downregulate BMPR2 expression in lymphocytes and lung vascular cells, which could amplify disease penetrance among BMPR2 mutation carriers and/or create a new risk factor among those without mutation [32, 58]. In animal model studies, Bmpr2 mutant mice develop PAH at a much higher level of penetrance upon exposure to 16-αOHE1 [33]. While estrogens reduced inflammatory cytokine expression acutely (a known beneficial effect of estrogens), they also amplified markers of pulmonary vascular injury . These included the alteration of genes related to thrombotic function, angiogenesis, planar polarity, and cellular metabolism [33]. Similarly, in a serotonin transporter (SERT) model of pulmonary hypertension known to amplify penetrance with female sex (in hypoxia, female mice that overexpress the serotonin transporter (SERT; SERT+ mice) exhibit PAH and exaggerated hypoxia-induced PAH, while male SERT+ mice do not [59, 60]); antagonism of ERα improves serotonin-dependent pulmonary hypertension and increases BMPR2 expression. In addition, ERα was found to be highly expressed pulmonary artery smooth muscle cells derived from PAH patients and increased in vitro via serotonin [61].
In contrast to the Bmpr2 and SERT murine data presented above, most animal models of pulmonary hypertension demonstrate a protective effect associated with female sex which appears to be mediated via parent compound estrogens, such as estradiol . The use of animal models to study pulmonary hypertension has been an area of controversy for many years. Admittedly, there is no perfect model and each animal model has advantages and drawbacks.
In most rodent models , including those which employ hypoxia and monocrotaline, and swine models (e.g., hypoxia), exposure to the insult of interest causes pulmonary hypertension to a much greater degree in male animals than females. The reasons for this are presently unclear [62–65]. Further complicating this, Sprague–Dawley rats exposed to monocrotaline develop pulmonary hypertension, but this is prevented and reversed with estradiol [66, 67]; however, ovariectomy of females promoted a pulmonary hypertensive phenotype [68]. Of particular interest given the data noted above that ERα promotes PAH, the beneficial effects of estrogen using the monocrotaline model appear to be mediated via ERβ specifically [66]. Further studies in additional animal models of PAH will be beneficial to help elucidate the complicated role of all sex hormones in the pathogenesis of PAH and explain the discrepancy between observations in humans and these animal models on the salutary or negative effects of estrogens.
The study of androgenic compounds , such as DHEA, is a logical consideration for pursuit using both animal models and cell-based studies. However, this is an area in need of expanded research. Some experimental pulmonary hypertension studies using male rates exposed to hypoxia and/or monocrotaline have shown improvement in the pulmonary hypertensive phenotype due to DHEA, and more studies are under way [43, 69, 70].
Conclusion
The female sex is associated with a higher incidence and prevalence of PAH in humans. The enhanced prevalence, and other recent findings, suggests that the female RV may be better suited to handle pulmonary hypertension for longer. But, the causes for this remain elusive. While sex hormones are increasingly implicated in the causation of PAH, the precise mechanisms require further study. Ultimately, precise understanding may result in important progress in solving this devastating disease via the development of novel therapeutic targets and prevention measures.
Sources of Support and Conflicts of Interest
This work was supported in part by NIH grant K23 HL 098743 (PI: EDA). The authors have no potential conflicts of interest to disclose.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


