Fig. 2.1
Simplified overview of sex hormone steroidogenesis . Synthesis of progestogens (blue), androgens (green), and estrogens (yellow) from cholesterol (red). Enzymes are shown in orange boxes. Abbreviations: 3β-HSD 3-beta-hydroxysteroid dehydrogenase, 17β-HSD 17-beta-hydroxysteroid dehydrogenase, CYP19A1 aromatase, CYP cytochrome, DHEA dehydroepiandrosterone, DHEA-S dehydroepiandrosterone-sulfate, DHT dihydrotestosterone, SULT2A1 sulfotransferase 2A1
Table 2.1
Circulating sex hormone levels in the human adult
Men <70 years | Men >70 years | Follicular | Preovulatory | Luteal | Pregnant women | Postmenopausal women | |
|---|---|---|---|---|---|---|---|
Estradiol | 15–50 pg/ml | n/a | 20–100 pg/ml | 150–400 pg/ml | 60–200 pg/ml | 10,000–40,000 pg/ml | 10–30 pg/ml |
Progesterone | 0.25–0.9 ng/ml | n/a | 0.3–1.2 ng/ml | 0.7–2.5 ng/ml | 1–18 ng/ml | 9–300 ng/ml | <0.2–1.1 ng/ml |
Testosterone | 2–15 ng/ml | 1.8–7.5 ng/ml | 0.2–0.8a ng/ml | 0.2–0.8a ng/ml | 0.2–0.8a ng/ml | 1–1.4 ng/ml | 0.2–0.8a ng/ml |
DHEA-S | 3470b ng/ml | 670 ng/ml | 2470a ng/ml | 2470a ng/ml | 2470a ng/ml | 2470a ng/ml | 450 ng/ml |
While circulating sex hormones have been implicated in the development and homeostasis of the normal lung [8], recent evidence has shown that sex hormones can also be synthesized in peripheral tissues such as the brain, skin, adipose tissue, bone, heart, and blood vessels [17, 43, 44]. In case of the latter, this occurs in both vascular endothelial cells (ECs) and smooth muscle cells (SMCs) [44, 45]. Important for the focus of this textbook, enzymes involved in sex hormone synthesis have been detected in the lung. Examples include aromatase (CYP19A1), 17β-HSD, CYP1B1, and CYP1A1 [8, 46–48]. This provides compelling evidence that not only does sex hormone-mediated modulation of the lung occur through the passive exposure to sex hormones in the systemic circulation but also that the lung has the capability to actively modulate sex hormone synthesis at the cellular level.
The Role of Biologically Active Estrogen Metabolites (Fig. 2.2)
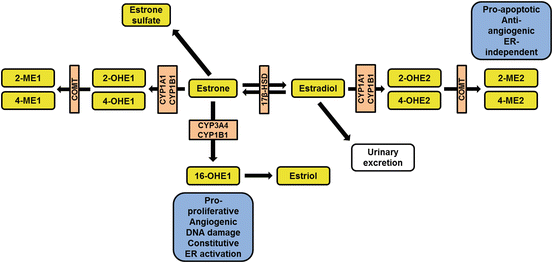
Fig. 2.2
Simplified overview of estrogen metabolism . Enzymes are shown in orange boxes. Abbreviations: 2-ME1 2-methoxyestrone, 2-ME2 2-methoxyestradiol, 2-OHE1 2-hydroxyestrone, 2-hydroxyestradiol (2-OHE2), 4-ME1 4-methoxyestrone, 4-ME2 4-methoxyestradiol, 4-OHE1 4-hydroxyestrone, 4-OHE2 4-hydroxyestradiol, 16-OHE1 16-alpha-hydroxyestrone, 17β-HSD 17-beta-hydroxysteroid dehydrogenase, COMT catechol-O-methyltransferase, CYP cytochrome P450
Three major estrogens are produced during steroidogenesis: estrone (E1), estradiol (E2), and estriol (E3). E2 is the primary circulating estrogen during reproductive years, whereas E1 is the primary circulating estrogen during and after menopause. E3 is primarily expressed during pregnancy and is the dominant circulating estrogen at that time [40]. Subsequent metabolism of estrogens is mediated by members of the cytochrome P450 superfamily: CYP1A1, CYP1B1, CYP3A4, and catechol-O-methyltransferase (COMT) [49]. Several of the CYPs are highly expressed in the lung, in particular CYP1B1 and CYP1A1 [46–48]. CYP1A1 and CYP1B1 facilitate the oxidation of estrogen into 2-hydroxy- (2-OHE), 4-hydroxy- (4-OHE), and 16α-hydroxy-estrogens [50, 51]. 2-OHE and 4-OHE metabolites have a short half-life and are rapidly converted to the more stable and biologically active 2-methoxy-estradiol (2-ME2) and 4-methoxy-estradiol (4-ME2). Of these, 2-ME2 has been shown to have antiproliferative, anti-angiogenic, anti-inflammatory, and pro-apoptotic effects on target organs that are independent of estrogen receptors [49, 50, 52]. Conversely, studies have demonstrated 4-ME2 and 16α-OHE1 to have pro-proliferative and pro-inflammatory effects on target tissues. In contrast to 2-ME-2, 16α-hydroxy-estrone (16α-OHE1)-mediated effects appear to be due, at least in part, to the constitutive activation of estrogen receptors, particularly estrogen receptor (ER)β [53]. 16α-OHE1 has also been shown to induce DNA damage [54, 55], leading to an association between individuals producing more 16α-OHE1 and an increased risk for diseases characterized by DNA damage, inflammation, or hyper-proliferation such as cancer and pulmonary arterial hypertension [2, 56–60], whereas individuals producing more 2-ME2 may be protected from these diseases [61, 62].
Mechanisms of Estrogen-Mediated Signaling
There are three known estrogen receptors : two classical steroid hormone receptors, ERα and ERβ [63–65], as well as a third less well-characterized G-protein-coupled receptor, GPR30 [66]. These receptors are widely, but differentially, expressed throughout the body including the cardiovascular, respiratory, immune, central nervous, and skeletal systems. ER expression and activity in these compartments are regulated by sex, age, diet, and sex steroid concentration (reviewed below).
ERα and ERβ are coded by different genes located on chromosomes 6 and 14, respectively. Despite these differences in chromosomal location, ERα and ERβ exhibit strong homology in their DNA-binding domain sequences (97 %). Both ERs also contain structural similarities in their ligand-binding domain and in their AF1 domain, through which different protein binding partners interact. However, these domains are less homologous than the DNA-binding regions; the ligand-binding domains of ERα and ERβ are 60 % homologous, whereas the AF1 domains are only 18 % homologous. ERα and ERβ are localized to the cell surface membrane, cytoplasm, and nucleus, whereas GPR30 is localized primarily to the cell surface [67]. ER localization is dependent at least in part on which signaling mechanism pathway is activated [63, 66]. In general, there are four major signaling mechanisms through which ERs exert their effects [2, 63–65, 68, 69]. The classical or genomic pathway is a common mechanism shared by the steroid receptor superfamily, including progesterone and androgen receptors. Briefly, estrogen binds to cytoplasmic ERα or ERβ which leads to a conformation change of the receptor, the dissociation of regulatory proteins, dimerization with another estrogen–ER complex (either as homodimer with the same ER subtype [e.g., ERα–ERα] or as heterodimer with the other subtype [e.g., ERα–ERβ]), and translocation to the nucleus. Once localized to the nucleus, the estrogen–ER dimer binds to estrogen responsive elements (EREs ) on target genes where it then regulates transcriptional activity as a classical transcription factor. In a second pathway, the estrogen–ER dimer indirectly modulates transcriptional activity by interacting with other transcription factors. The third mechanism of ER activation occurs through an estrogen-independent mechanism. In this scenario, the ER is activated through phosphorylation by a non-estrogen growth factor (i.e., epidermal growth factor [71]). The ER then dimerizes, translocates to the nucleus, and regulates the transcription of target genes. The fourth mechanism is known as the non-genomic signaling pathway. In this pathway, estrogen binds to ER localized at the cell membrane. This ligand–receptor complex then trans-activates cytoplasmic protein kinases such as PI3K or MAPK [72]. Examples for non-genomic estrogen signaling include rapid effects on endothelial nitric oxide synthase (eNOS) or calcium channel activity in the vasculature [73, 79]. This mechanism of estrogen-mediated effects was initially described for GPR30, but recently ERα and ERβ have also been shown to mediate estrogen signaling through this pathway [72, 74]. The non-genomic pathway occurs rapidly (within seconds or minutes after the receptor is activated by estrogen) and is likely an important pathway in tissues where a rapid cellular response to injury is required, i.e., the cardiovascular system [75–79]. This is in contrast to signaling via genomic pathways, which occurs over several hours or even days [2, 69].
Regulation of Estrogen Signaling
Estrogen-mediated signaling is a highly complex process that is regulated in several ways [68]. Globally, estrogens are regulated through the differential cell-type and tissue-specific expression pattern of ERs [6, 8]. Sex, age, and the presence of health versus disease are major mediators of estrogen signaling [9, 43, 80]. ER expression is also temporally regulated based on the phase of menstrual cycle and by abundance of circulating estrogen levels [81–83]. Nutritional influences may also interfere with ER expression and/or activity. For example, indole-3-carbinol, a key ingredient of cruciferous vegetables, is a negative regulator of estrogen signaling [61]. On the other hand, xenoestrogens introduced into the environment through industrial, agricultural, and chemical processes exert pro-estrogenic effects [84, 85]. On a molecular level, estrogen signaling is modulated through heat shock protein chaperone interactions with ERs and through post translational modification of the receptors [86]. For example, ERs can be S-nitrosylated or acetylated, with the first modification inhibiting and the latter process enhancing the transcriptional activity of ERs [87]. Additionally, ER gene promoters themselves can be methylated, thus reducing ER mRNA levels [88]. Conversely, estrogen has been shown to upregulate ER transcription, while other sex hormones (e.g., progesterone) can negatively regulate the expression of ER mRNA [89, 90]. Other transcription factors , such as MEF2 and HDAC2, may inhibit ER signaling, and ubiquitination and proteasomal degradation decrease ER abundance. Lastly, several splice variants of ERα and ERβ are able to interact and positively or negatively modulate the activity of ERs, thus adding another layer of complexity to regulation of estrogen-mediated signaling [63, 91].
Progesterone and Androgen-Mediated Signaling
Progestogens are a class of steroid hormones characterized by their ability to bind to and activate progesterone receptors (PR ). Progesterone is the most abundant and biologically active progestogen . Other endogenous progestogens include 17-hydroxyprogesterone, 5α-dihydroprogesterone, and 11-deoxycorticosterone, all of which are converted to other intermediates in the steroidogenesis pathway [15, 22, 92]. As multiple cell and tissue types have the capacity to convert cholesterol to steroid hormones, progestogens are found in many cell types [93, 94].
There are several major androgens . These include testosterone and dihydrotestosterone (DHT), DHEA/DHEA-S, androstenediol, androstenedione, and androsterone. DHEA/DHEA-S and androstenediol are considered weak steroids and precursors for testosterone and estrogen synthesis [28]. However, there is increasing interest in the effects of DHEA/DHEA-S on the cardiopulmonary system [2], and androstenediol plays a role as a major regulator of gonadotropin secretion [95]. While testosterone and DHT exert multiple biologically relevant effects, they (together with androstenedione) also function as important estrogen precursors. Consequentially, androgen pathways and their receptors are expressed in a wide range of tissues throughout the body, including the cardiopulmonary system [96–98].
Progestogens a nd androgens mediate their effects on lung function in health and disease through two progesterone receptors (PR-A and PR-B) and an androgen receptor (AR) , respectively. ARs, used by testosterone and DHT, and PRs are found widely throughout the body, and both receptors are present in the human lung [93, 98], vascular ECs [99, 100], SMCs [101, 102], and the heart [97, 103], suggesting relevant physiologic effects in these organs. In general, AR and PR signaling occurs in a similar manner to ER signaling, via genomic or non-genomic pathways . Vasodilatory effects of testosterone, however, appear to be independent of the AR [104]. Little is known about receptor signaling pathways employed by DHEA in the cardiopulmonary system, and no specific DHEA receptor has been identified. While DHEA actions appear to be independent of ERs or ARs [105], DHEA has been reported to bind to a specific EC membrane receptor coupled to G-alpha i2,3 with subsequent activation of eNOS [106]. In addition, DHEA stimulates Akt/eNOS signaling through activation and/or upregulation of sigma-1 receptor. Non-genomic DHEA signaling has been described as well [69].
Regulation of Progesterone and Androgen Signaling
Similar to ERs, PRs and AR regulate steroid hormone effects through cell- and tissue-type-specific expression patterns , which are modulated by sex, age, sex hormone concentrations, and menstrual cycle [107, 108]. Like ERs, PRs and ARs are regulated by chaperone proteins and by degradation via the proteasome. Both receptor types can also be posttranslationally modified to enhance or repress transcriptional activity of target genes. Progesterone plays a major role in regulating expression of the PRs. PR-B is the primary activator of progesterone-mediated gene transcription, whereas PR-A exerts inhibitory effects and can act as a repressor of PR-B signaling. In addition to progesterone, estrogen regulates PR expression, indicating significant cross-talk between ER and PR pathways. For example, the presence of estrogen may enhance the effects of PR-B, whereas the presence of androgens or PR-A can inhibit estrogen and progesterone pathways, again demonstrating the importance of sex hormone balance to maintain tissue homeostasis [92, 109–111].
Sex Hormones and Lung Development
Sex Hormones and the Prenatal and Early Postnatal Lung
Sex hormones have been shown to modulate lung development as early 16–24 weeks of gestation [8, 112]. The lung itself continues to develop after birth until 2–4 years of age and continues to change in complexity until early adolescence [8, 113–116]. During neonatal lung development, androgens inhibit surfactant production in the developing neonates, causing delayed surfactant production and delayed alveolar maturation in male neonates compared to female neonates [114, 117]. Additionally, androgens enhance lung airway branching leading to the development of larger lungs in male neonates [8, 117, 118]. These two roles of androgens in the developing lung are clinically relevant, as male neonates exhibit an increased incidence and mortality of infant respiratory distress syndrome, a condition that mainly results from surfactant deficiency and incomplete fetal lung maturation [112, 114, 117]. Mechanisms of androgen-mediated enhancement of airway branching and inhibition of surfactant production appear to involve upregulation of epidermal growth factor (EGF) and transforming growth factor (TGF)-β pathways . However, while this has been demonstrated in vitro and in animal models, it remains unclear if these mechanisms are also responsible for surfactant inhibition in humans [119–121].
The fetal lung also contains ERs , which have been implicated in alveolar development [112, 118, 121]. For example, genetic deletion of ERα or ERβ in mice decreases the number and increases the size of alveoli; these changes are more pronounced in female mice [120]. These ER-mediated effects on alveoli size are likely mediated through effects on platelet-derived growth factor (PDGF) signaling [122]. Morphologically and structurally, compared to male lungs, female lungs are smaller and have fewer respiratory bronchioles. The larger lung size of males at any given age results in an increase in the number of alveoli and alveolar surface area as compared to females; however when normalized for unit area and lung volume, the number of alveoli, at least at birth, is not different between sexes [6, 112, 118].
Additional evidence that steroid hormones regulate fetal lung development comes from studies of the expression of steroidogenesis enzymes such as 17β-HSD, 3β-HSD, 5α-reductase, and aromatase in human fetal lung tissue [8, 122]. Although these studies strongly suggest active modulation of sex hormone pathways in the human fetal lung, it is unlikely that de novo synthesis of sex hormones from cholesterol occurs during this time because CYP17A1 (the enzyme responsible for converting progestogens to androgens) is not detectable [123]. Thus, the presence of several steroidogenesis enzymes suggests that the conversion of precursors into biologically active sex hormones occurs locally in the prenatal and early postnatal lung and that these hormones play a role in mediating lung development.
Taken together, the data reviewed in this section suggest a significant role for estrogens and androgens in lung development, implying them in alveolarization and surfactant production.
Sex Differences in the Pubescent Lung
Female airways and lung parenchyma grow proportionally during childhood and puberty; in males, however, airway growth lags behind the development and growth of other lung structures, resulting in fewer alveoli for the number of airways as well as narrower airways, a phenomenon called dysanapsis [115, 116, 124, 125]. This disproportional growth of lungs and airways results in higher airway resistance in males [112]. On the other hand, males have larger conducting airways than females throughout adolescence and into adulthood. During adolescence, males have higher respiratory pressures compared to females. This difference is attributed to the role of testosterone in changing the shape of the thorax and respiratory muscles during puberty [126]. Other sex differences observed in neonates and children are maintained through puberty, with the dysanaptic growth of the airways in males maintained into adulthood .
Sex Differences in the Aging Lung
Sex differences also occur in the aging lung. During the aging process, there is a decrease in elastic recoil of the lung and large airways as well as of alveolar air volume, whereas abundance of connective tissue increases [127, 128]. However, these changes appear to be less pronounced in females compared to males and occur at a later age [129]. Menopause has significant effects on lung function. For example, during menopause, which is associated with low levels of progesterone and estrogen, women are less likely to develop asthma compared to men [3]. Furthermore, the aging-mediated associated decrease in sex hormone production negatively affects alveolar fluid clearance [130]. Finally, loss of anabolic effects of sex steroids on muscle structure and function during aging is associated with a generalized loss of muscle mass; this results in changes in chest wall architecture and a general loss of respiratory muscle strength [131].
Taken together, the reviewed sex differences during lung development and throughout aging illustrates how minor changes in lung structure and development can have a major impact on health in later life. Despite emerging evidence of the role of sex hormones in regulating lung development, architecture, and function, further understanding of these processes is required to determine their role in aging .
Effects of Sex Hormones on the Healthy Lung
Nearly every major cell type of the lung is affected by sex hormones. This section will review these effects using a compartment-based approach that focuses on bronchial and alveolar epithelium, airway smooth muscle cells, lung parenchyma, the pulmonary vasculature, as well as lung immune cells, concluding with a brief review of sex hormone effects on respiratory mechanics and respiratory drive. Table 2.2 provides a synopsis of the reviewed data.
Table 2.2
Sex hormone effects on major cell types of the lung
Pulmonary cell type | Estrogen | Progesterone | Androgen |
|---|---|---|---|
Bronchial epithelium | ↑ Nitric oxide generation | ↑ Nitric oxide generation | Unknown |
↑ Cell proliferation | ↓ Cell proliferation | ↑ Cell proliferation | |
Alveolar epithelium | ↑ Alveolar regeneration and development | Unknown | ↓ Alveolar maturation |
↑ Alveolar fluid clearance | ↑ Alveolar fluid clearance | ↓ Surfactant production | |
↑ ENaC expression | ↑ ENaC expression | Unknown | |
Airway smooth muscle | ↓ Intracellular calcium relaxation | ↓ Intracellular calcium relaxation | ↓ Intracellular calcium relaxation |
↑ Cell proliferation | ↓ Cell proliferation | Unknown | |
Parenchyma (ECM/fibroblasts) | ↓ Fibroblast proliferation | Fibroblast apoptosis | Unknown |
↑ Laminin/fibronectin | Unknown | ↓ Laminin/fibronectin | |
↓ MMPs | ↓ MMPs | ↑ MMPs | |
Pulmonary artery smooth muscle | ↓ Intracellular calcium relaxation | ↓ Intracellular calcium relaxation | ↓ Intracellular calcium relaxation |
↑ Cell proliferation | Unknown | Unknown | |
↓ Proliferation at supraphysiological concentrations | Unknown | Unknown | |
Pulmonary artery endothelium | ↑ Nitric oxide generation | ↑ Nitric oxide generation | Unknown |
↑ Cell proliferation | Unknown | Unknown | |
↓ Proliferation at supraphysiological concentrations and during hypoxia | Unknown | Unknown | |
Immune cells | Th2 profile | Th2 profile | Th1 profile |
↑ Mast cell degranulation | ↓ Mast cell degranulation | ↑ Mast cell degranulation | |
↑ Eosinophil degranulation | Unknown | Unknown |
Bronchial Epithelium
The bronchial epithelium functions as a barrier between the body and external pathogens , allergens, and pollutants as well as a modulator of airway tone [132]. Inflammation of the bronchial epithelium is a key step in the pathogenesis of a number of diseases including asthma and chronic bronchitis [133]. Likewise, the bronchial epithelium is the starting point for several lung cancers [134]. Despite the importance of the bronchial epithelium, little is known about the role of sex hormones in this tissue .
Both ERs are expressed in bronchial epithelial cells (BECs ). Whereas in immortalized airway epithelial cell lines ERβ is expressed more abundantly, in primary BECs both ERs are expressed at similar levels [135, 136]. These data emphasize that results derived from studies of ER expression in immortalized cells cannot be extrapolated to primary cells or the in vivo scenario. Expression of PR or AR in the bronchial epithelium has not been studied.
Nitric Oxide (NO) Signaling
One mechanism by which estrogens modulate BECs is through the regulation of NO signaling. The main source of NO production in the airways in response to inflammation is through inducible NO synthase (iNOS) [137, 138]. However, endothelial NOS (eNOS) also plays a major role in the bronchial epithelium through the regulation of airway blood flow [139]. NO generated as a result of BEC iNOS and eNOS can be quantified as exhaled NO (eNO) and is used clinically as an indicator of airway inflammation [140]. iNOS and eNOS facilitate the conversion of l-arginine to l-citrulline and thus the release of NO, a by-product of this reaction and a potent bronchodilator [137]. Studies in the vascular endothelium have shown that estrogens facilitate the dissociation of inactive eNOS from caveolae, leading to eNOS activation and a subsequent increase in NO production and vasodilation [141, 142]. Whether the same phenomenon also occurs in BECs is unknown. However, what is known is that in immortalized BEC lines, E2 acutely increases eNOS activity [135]. This is inhibited by the ER antagonist, fulvestrant, suggesting an ER dependence of effects. Similarly, activation of ERα or ERβ with selective receptor agonists in primary BECs results in eNOS phosphorylation and increased NO production [143]. This effect occurs within minutes, strongly implicating a role of non-genomic estrogen signaling in NO production in the bronchial epithelium. Additional evidence from isolated human female bronchial rings shows that physiological concentrations of estrogens are sufficient to produce a robust bronchodilator response. Denuding the epithelium blunts this effect, suggesting that estrogens non-genomically induce NO production in the bronchial epithelium and thus modulate bronchodilation [143]. This mechanism is remarkably similar to what has been described in the vascular endothelium, where estrogen-mediated non-genomic stimulation of eNOS in endothelial cells results in vasodilation [78].
Exhaled NO production in humans may fluctuate with menstrual cycle status; however there are several studies with conflicting results. For example, one study showed that eNO levels decreased when estrogen levels were high and increased when progesterone levels were high [140]. Women taking an oral contraceptive did not have such fluctuations in eNO production [144, 145]. On the other hand, data also exists indicating that the menstrual cycle does not significantly affect NO levels [81, 146].
BEC Proliferation
In addition to mediating bronchodilator responses in BECs, estrogens may affect BEC proliferation. While sex hormones are known to regulate cell proliferation pathways in other tissues and cell types [147, 148], data on estrogen-mediated proliferative responses in BECs are sparse. While in some lung adenocarcinoma cell lines pro-proliferative responses were mediated through ER activation, this did not occur in all cell lines studied [136]. In particular, lung adenocarcinoma cell lines from female, but not male, patients appear to be responsive to pro-proliferative effects of E2, an effect that was linked to differences in nuclear localization of ERα. Effects of other sex hormones on BECs remain uncharacterized.
Mucus Clearance
Lastly, E2 has been shown to regulate chloride secretion, implicating estrogens in a role for susceptibility to infection and impaired mucus clearance in the bronchiolar epithelium [82]. This role has particular significance for female cystic fibrosis patients, who have cyclical decreases in lung function correlating with menstrual cycle [82]. Taken together, these data suggest estrogens as modulators of BEC NO production, proliferation, and chloride secretion. Effects of other sex hormones and expression patterns and roles of AR and PR remain poorly characterized.
Alveolar Epithelium
The alveolar epithelium consists of type I cells which are squamous epithelial cells comprising the structure of the alveolar wall and type II cells which are cuboidal epithelial cells responsible for producing and secreting surfactant. ERs, PRs, and AR have been detected in alveolar epithelial cells (AECs) of normal lung tissue and in cell lines [98, 149]. Several steroidogenesis enzymes, including 17β-HSD and aromatase, are found in AECs, indicating that sex hormone precursors may be modified to biologically active sex hormones locally [150]. However, as with the bronchial epithelium, most studies in AECs have focused on the role of sex hormones in the context of disease. It is thus difficult to extrapolate a role for sex hormone signaling and function in the healthy alveolar epithelium.
Surfactant Production, Alveolar Development, and Regeneration
One area where the role of sex hormones in the healthy lung is relatively well defined is the role of androgens in delaying maturation of type II AECs and surfactant production in the prenatal lung [151]. Here, androgens inhibit surfactant production through modulation of EGF and TGF-β pathways (which are positive and negative regulators of lung maturation, respectively [119–121]). In fact, an androgen-induced delay in EGF receptor binding during lung development has been described in rabbits [152]. Alternatively, exposure of female fibroblasts to DHT results in a dose-dependent increase of TGF-β1 activity [119].
Estrogens modulate AECs through multiple mechanisms. For example, E2 regulates type I AEC development and regeneration [153, 154]. In mouse studies, ovariectomy (OVX) of sexually immature wild-type female mice results in a decrease in the number of alveoli during development and lung maturation. Treatment with E2, on the other hand, restores alveolar numbers in OVX mice [154]. Furthermore, ERα or ERβ knockout mice exhibit similar alveolar developmental defects, demonstrating that both ERα and ERβ are required for the normal development of pulmonary alveoli in female mice [154]. OVX of wild-type adult female mice results in the loss of 40 % of alveoli. E2 treatment induces alveolar regeneration through a distinct ERα-dependent mechanism and restores alveoli number and lung volume [153]. These data thus demonstrate a role for estrogen-mediated signaling in alveolar development and regeneration and provide a mechanism for increases in FVC and FEV1 observed in postmenopausal women undergoing hormone replacement therapy [155].
Alveolar Fluid Homeostasis
Sex hormones also regulate alveolar fluid homeostasis in healthy AECs. Estrogens and progestogens (specifically E2 and progesterone) have been identified as key mediators of alveolar fluid clearance, a process that occurs through regulation of epithelial sodium channel (ENaC ) expression , abundance, and activity [130, 156, 157]. ENaCs are essential for sodium transport and fluid balance in the alveolar space. In particular, fluid is absorbed into AECs through ENaCs and extruded from the cell through Na/K+ ATPases [158]. These channels are often dysregulated in pulmonary diseases such as cystic fibrosis and acute respiratory distress syndrome (ARDS ) , leading to abnormal fluid clearance of the alveolar space [159]. Interestingly, aging and the associated decrease in sex hormone production can lead to deficiencies in alveolar fluid clearance [130]. On the other hand, E2 treatment of adult type II AECs causes, through a GPR30-dependent mechanism, an increase in ENaC abundance and localization to the apical membrane [130]. Similarly, treatment of fetal type II AECs with progesterone leads to an increase in ENaC expression and activity [156].
Airway Smooth Muscle
The airway smooth muscle (ASM) is responsible for modulating the tone of the bronchial airways by constricting or dilating the airway. The role sex hormones in the regulation of ASM function is better understood than their role in other lung compartments, in part due to increasing interest in the investigation of sex differences in asthma and COPD.
Regulation of Airway Tone
In general, estrogens exert bronchodilatory effects , but time- and dose-dependent differences seem to exist. These effects are mediated through both epithelium-dependent and epithelium-independent mechanisms leading to decreased intracellular Ca2+ levels. Chronic administration of low doses of E2 (10 μg/kg) increases the concentration of inhaled acetylcholine (ACh) necessary to double airway resistance in OVX rats [160, 161]. On the other hand, OVX rats treated with high doses of E2 (100 μg/kg) exhibited increased responsiveness to ACh-induced vasoconstriction [161]. Together, these data suggest a dose-specific effect of E2 on ASM responses to ACh. Studies in rabbit tracheal strips pre-constricted with ACh showed that treatment with 100 μM E2 was able to relax the airway smooth muscle layer in an epithelium-independent manner [162, 163]. E2-mediated effects were found to be due in part to increased prostaglandin synthesis with downstream increases in cAMP levels, as well as through increases in cGMP signaling, both leading to attenuation of muscle tone through inhibition of Ca2+ influx [163].
In addition to indirectly regulating Ca2+ concentrations via cAMP and cGMP, estrogen is also able to directly modulate Ca2+-mediated responses via effects on Ca2+ channels [164]. For example, in human ASM cells, physiologically relevant concentrations of E2 (100 pM–10 nM) substantially decrease Ca2+ responses to histamine through an ERα-mediated mechanism [165]. E2 effects on ASM are regulated, in part, through decreased Ca2+ influx through regulation of L-type channels and store-operated entry [165]. Taken together, these studies indicate that estrogen-mediated bronchodilation in ASM occurs via reduction of intracellular Ca2+ concentrations through ER-dependent mechanisms.
The effects of progesterone on ASM tone are less well defined. Progesterone potentiates the dilatory effect of isoprenaline in constricted pig bronchial rings [166]. In these studies, progesterone, although administered at a high dose (40 μM), had a more robust dilatory effect than testosterone, but was less effective than estradiol. Progesterone also prevents histamine- or carbachol-induced contraction in guinea pig trachea [167]. Progesterone-mediated regulation is proposed to occur through direct inhibition of Ca2+ influx [168]. Combined, these studies provide evidence for a bronchodilator effect of progesterone on ASM similar to, but not as robust as, that of estrogen.
Similar to estrogen and progesterone treatment, testosterone treatment causes a dose-dependent bronchodilator effect on ASM, as shown in pre-constricted rabbit tracheal strips [162]. However, unlike estrogen and progesterone treatment, whose bronchodilatory effects are both epithelial dependent and independent, removal of the epithelium in testosterone-treated tracheal strips completely attenuates the dilatory response, suggesting testosterone-mediated relaxation is epithelial dependent. Interestingly, inhibition of AR with flutamide does not alter the dilatory effects of testosterone, suggesting an AR-independent mechanism of action. Furthermore, dilatory effects were maintained after administration of bovine serum albumin (BSA)-conjugated (and thus cell membrane-impermeable) testosterone, a phenomenon consistent with non-genomic signaling [162]. Similar to the previous study, removal of the epithelium attenuated the testosterone response. Taken together, these studies suggest that testosterone is mediating bronchodilator effects through non-genomic interaction at the cell surface with the AR receptor. These bronchodilator effects are mediated through voltage-gated Ca2+ channel antagonism .
Of clinical relevance, in asthma, a disease characterized by both inflammation and hyperresponsiveness of the airway, there is greater prevalence and morbidity in adult women compared to men [169, 170]. Women also have more severe and more frequent asthma exacerbations than men [113], with 40–50 % of women having cyclic variations and exacerbations during the premenstrual/late luteal phase of their menstrual cycle [171]. At this point in the menstrual cycle, estrogen levels are at their lowest, and progesterone levels are increased [39, 172]. Studies in healthy volunteers and asthma patients demonstrate that the luteal phase of the menstrual cycle is characterized by the lowest FVC and FEV1, suggesting a direct effect between sex hormone levels and lung function [81]. Along these lines, postmenopausal women exhibit decreased airway function [173], and postmenopausal asthma patients are characterized by more severe disease compared to men and reproductive-aged women [173]. Together, while the exact mechanism is unclear, these studies suggest that sex hormones modulate airway tone and asthma prevalence and severity [3, 81]. However, these relationships are complex, as there is also evidence that administration of estrogen to menopausal women is associated with increased rates of newly diagnosed asthma [174]. Disparate effects on airway tone vs. airway inflammatory responses and ASM proliferation may contribute to these complex relationships. Environmental effects on ERs and estrogen metabolism may also play a role (e.g., hyperoxia) [48].
ASM Proliferation
Evidence also exists that sex steroids, in addition to affecting ASM tone, exert physiologically relevant effects on ASM proliferation. For example, E2 has been shown to enhance proliferation of ASM cells [175]. Such effects have been linked to worsening effects of estrogens on airway remodeling in asthma [176]. DHEA, on the other hand, inhibits rat tracheal smooth muscle cell proliferation after stimulation by fetal bovine serum or PDGF through inhibition of activator protein-1 [177]. However, more detailed exploration of the role of sex hormones on ASM proliferation is required; such studies would help to further decipher the role of these hormones in airway diseases such as asthma .
Parenchyma
For the purpose of this chapter, the lung parenchyma refers specifically to the alveolar tissue with respiratory bronchioles, alveolar ducts, and terminal bronchioles. The parenchyma provides structural support, participates in gas exchange, and is the site of extracellular signaling and regulation. The role of sex hormones on extracellular matrix deposition and modulation of enzyme regulators of the extracellular matrix is of clinical interest since men are more likely to develop and die from idiopathic pulmonary fibrosis than women [112]. Studies in the bleomycin model of pulmonary fibrosis, however, suggest female sex hormones as being pro-fibrotic [178]. Along those lines, female rats treated with bleomycin developed more severe fibrosis than males. OVX female rats, on the other hand, had less severe fibrosis compared to intact females, and E2 replenishment restored fibrosis levels to those of intact females [178]. In vitro studies suggest that the pro-fibrotic effect of E2 is likely mediated through upregulation of procollagen 1 and TGF-β1 gene expression in fibroblasts [178]. Studies in the mouse bleomycin model of pulmonary fibrosis, however, suggest a greater degree of fibrosis in male animals [179]. Here, castration of male mice is protective against the development of pulmonary fibrosis, while DHT replenishment attenuates this protective effect [179]. Taken together, even though discrepant with regard to protection versus harm, the reported in vivo and human data suggest sex hormones as potential modulators of fibrotic processes. Effects on proliferation may also play a role in this process. For example, high concentrations (5 μg/ml) of E2, testosterone, or progesterone decrease proliferation of human fetal lung fibroblasts [180, 181]. Furthermore, decreased proliferation was noted in lung myofibroblasts treated with E2, mediated through non-genomic signaling via activation of the Raf1–ERK–MAPK pathway [181]. However, the relevance of these in vitro studies to the in vivo scenario remains unknown.
Collagen, fibronectin, laminin, and other components of the extracellular matrix of the parenchyma are important for the structural integrity and regulation of lung homeostasis, and their dysregulation can result in various pathologies. However, effects of sex hormones on extracellular matrix components have only recently been explored and have yet to be studied in the lung. Studies using mouse skin biopsies have shown that adult male mice have 25 % more lung hydroxyproline (representative of collagen deposition) present than age-matched females [182]. AR knockout mice did not have such an increase in hydroxyproline, implicating male sex hormones as potential modulators of collagen deposition in the parenchyma of the lung [182]. Despite these studies, the exact mechanisms of sex hormone regulation of the lung parenchyma are mostly unknown.
Pulmonary Vasculature
Due to the well-described gender differences in pulmonary arterial hypertension (PAH ) [2, 70, 73, 183–185], there has been increasing interest in the study of sex hormone effects on the pulmonary vasculature. While a significant body of knowledge has been derived from specific study of the pulmonary vasculature, other data have been extrapolated from studies of systemic vascular cells. Studies from our laboratory demonstrate that both ERα and ERβ are expressed in pulmonary artery endothelial cells (PAECs ) as well as smooth muscle cells (PASMCs ) [186, 187] (Fig. 2.3 ). In addition, enzymes critical for estrogen synthesis and metabolism have been detected in lung blood vessels; these include aromatase and CYP1B1 [46, 47], suggesting that local estrogen production and metabolism occur in the pulmonary vasculature. This is of importance, as alterations in estrogen synthesis have recently been linked to PAH pathogenesis [2, 188]. GPR30, while present in the lung (Lahm, Frump; unpublished), has not yet been described in the pulmonary vasculature, but has been found in systemic vascular cells [189]. Similarly, AR and PR have been detected in vascular ECs [99, 100] and SMCs [101, 102].
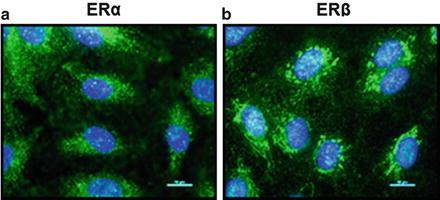
Fig. 2.3
ERα and ERβ are expressed in human pulmonary artery endothelial cells (HPAECs). Representative immunocytochemistry images of cultured HPAECs. Expressions of ERα (green, a) and ERβ (green, b) are shown. Note that localization of both ERs correlates with expression in cytoplasm and nucleus. Cell nuclei are stained with DAPI (blue). Images are 40x; scale bar indicates 10 μm
Estradiol Effects on Vasomotor Tone
In general, sex hormones serve as vasodilators of the vasculature, with most data being available for E2. Multiple pathways have been implicated in E2’s vasodilator effects. Non-genomic stimulation of eNOS appears to be the most prominent mechanism. In particular, in PAECs, estrogen acutely activates eNOS with subsequent production of NO, and both ERs have been implicated in these effects via two different non-genomic signaling mechanisms [190, 191]. While such effects have been demonstrated in various cell types as well as in isolated pulmonary artery (PA) rings, various downstream mediators have been implicated in these findings. For example, in transformed rat lung vascular ECs, eNOS activation was mediated through an ERα- and Akt-dependent mechanism [192, 193]. In PAECs, on the other hand, estrogen-mediated eNOS activation occurred through ERβ localized to caveolae [76]. In isolated PA rings from healthy rats, both ERs have been implicated in E2’s non-genomic vasodilator effects, with ERα attenuating potassium-induced vasocontraction and ERβ attenuating hypoxic vasoconstriction (HPV) [194]. These effects are mediated in an endothelial cell- and NO-dependent manner. Such effects are physiologically relevant, as PA rings from proestrus rats exhibit less HPV than arteries from animals with lower circulating E2 levels, such as estrus or diestrus rats as well as male rats [83].
E2 also targets other vasoactive mediators. For example, in PAECs, E2 attenuates hypoxia-induced upregulation of the vasoconstrictor endothelin-1, an effect mediated by interfering with activity of hypoxia-inducible factor (HIF)-1α [195]. Lastly, E2 enhances prostacyclin synthase activity and prostacyclin production through an ERβ- and calcium-dependent mechanism and also regulates the upstream mediators, cyclooxygenase-1 and cyclooxygenase-2 [73].
While many of E2’s PAEC effects ultimately affect PASMC tone, direct effects on PASMCs may also contribute to E2’s pulmonary vasodilator effects. However, such data mostly stem from the systemic circulation, where E2 has a vasodilatory effect on SMCs. For example, E2 hyperpolarizes calcium-dependent potassium channels (BKCa) in a cGMP-mediated manner, thus leading to SMC relaxation [196]. Estrogens are also purported to promote SMC relaxation through inhibition of the Rho kinase pathway [197]. Despite robust evidence for the role of estrogen on systemic vascular SMCs, there is little direct evidence for the role of estrogens on PASMCs, although such effects are hypothesized [198]; consequentially, this still represents a knowledge gap in the field.
Estradiol Effects on PA Cell Proliferation
In contrast to the small number of studies evaluating vasorelaxor effects of estrogens on PASMCs, multiple studies have evaluated E2 effects on PASMC proliferation. The observation that ERs, aromatase, and CYP1B1 are expressed in PASMCs indicates that conversion of sex hormone precursors into biologically active estrogens and metabolites may be regulated locally in PASMCs [186–188]. Many of the currently published investigations identified estrogens as inducers of PASMC proliferation [201, 202]. However, in hypoxic PASMCs, the opposite seems to be the case, and E2 has been identified in this context as an inhibitor of PASMC proliferation [203, 204]. Interestingly, PASMCs isolated from proestrus rats exhibited less proliferation under hypoxia conditions than cells from estrus or male rats [204]. Mitogenic effects of E2 also seem to be dose dependent, as at least one study suggests a shift toward anti-mitogenic effects on PASMCs at higher concentrations [205]. Similar dose-dependent effects on proliferation have also been described for PAECs, with physiological concentrations promoting, but supraphysiological concentrations inhibiting, proliferation [205]. Our laboratory found that proliferative E2 effects on rat PAECs are context dependent, with pro-proliferative effects being detected in room air conditions, but antiproliferative effects being observed during hypoxia (1 % O2) [206]. Similar hypoxia-specific effects were seen when evaluating ERK1/2 activation and VEGF secretion, both of which were attenuated in hypoxia, but not normoxia. Along these lines, expression of the cell cycle inhibitor p27kip1 was decreased by E2 at room air, but enhanced during hypoxia. These effects appear to be mediated in an ER-dependent manner, with effects on ERK1/2 activation being attenuated by both ERα and ERβ blockades, thus implicating both ERs in these effects. Hypoxia-induced upregulation of ERβ PAEC expression may mediate such hypoxia-specific effects (Fig. 2.2) [186, 187]. Biologically active effects of estrogen metabolites also seem to regulate PASMC and PAEC proliferation, with 2-ME acting as a mitogen [205], while 16α-OHE1 appears to mediate pro-proliferative signaling [46].
Taken together, the current body of literature suggests context-dependent E2 effects on PAEC and PASMC proliferation, with antiproliferative effects predominantly being seen during hypoxia and/or at supraphysiological concentrations. Genetic alterations (e.g., BMPR2 mutations) and environmental exposures (e.g., drugs interfering with serotonin signaling, such as appetite inhibitors) may affect such responses .
E2 Effects on Angiogenesis
Cyclic angiogenic changes occur in the lung parallel with cyclic gas exchange changes that occur during the menstrual cycle [207]. Specifically, over the course of their menstrual cycle, healthy women exhibit variable lung diffusion capacity, with the nadir occurring during the follicular phase. This decrease in lung diffusion capacity was accompanied by a 25 % decrease in vascular capillary blood volume and a 24 % decrease in CD34+ CD133+ circulating endothelial progenitor cells [207]. Expression of the pro-angiogenic cytokine stem cell factor (SCF ) correlated with these changes; however vascular endothelial growth factor (VEGF) did not. Interestingly, OVX mice with estrogen replenishment exhibited an increased number of microvessels compared to OVX mice without estrogen replacement [207]. This suggests that pulmonary vascular angiogenesis is regulated by estrogen and that lung microvascular density and diffusion capacity vary throughout the menstrual cycle, thus paralleling a pattern that has been described previously in uterine endometrium [208, 209].
Effects of Progesterone, Testosterone, and DHEA
The effects of progesterone on the pulmonary vasculature are less well characterized. In a study of isolated PAs, progesterone had the strongest vasodilatory effect compared to estrogen and testosterone [210]. This vasodilatory effect of progesterone treatment was due in part to endothelial-dependent NO mechanisms. Treatment with an NOS inhibitor reduced progesterone-mediated vasodilation. Additional study demonstrated that the vasodilatory effects of progesterone were also mediated through inhibition of voltage-gated and receptor-mediated Ca2+ channels [211]. Further mechanisms of pulmonary vasomotor tone regulation by progesterone are largely unknown.
rone-mediated effects in the pulmonary vasculature are not well studied. Two studies identified testosterone as a more potent vasodilator of isolated PAs than E2, with PAs from male rats being more sensitive to the effects of testosterone than those from female rats [104, 210]. Testosterone’s vasodilator effect is mediated at least in part through inhibition of voltage-gated and receptor-mediated Ca2+ channels, while AR, NO, or prostaglandins do not appear to be involved [104]. Such vasodilator effects were recapitulated in human PAs and isolated perfused human lungs, but no sex differences were noted in this context [212]. Further studies demonstrated that most robust vasodilator effects of testosterone were observed at physiological concentrations and only in the presence of an intact endothelium [213].
DHEA effects in the pulmonary vasculature are slightly better understood. Several studies identified DHEA acts as a pulmonary vasodilator. In isolated ferret lungs preconstricted with the K+ inhibitor tetraethylammonium, DHEA treatment, through a cAMP- and cGMP-independent mechanism, stimulated the opening of K+ channels and decreased pulmonary artery pressure [214]. In another study using isolated rat lungs and PAs, DHEA treatment, through a prostaglandin-, endothelial-, NO-, and cGMP-independent mechanism, opened voltage-gated K+ channels leading to the relaxation of PAs. The mechanism is believed to be through reduced NADPH expression and a change in PASMC redox status [215]. One specific mechanism by which DHEA is thought to regulate vasodilation is through its role as a noncompetitive inhibitor of glucose-6-phosphate dehydrogenase [216]. This, in turn, inhibits the pentose phosphate pathway and NADPH production, changing the redox state of the cell. This mechanism was also linked to potential antiproliferative effects of DHEA in the pulmonary vasculature [217]. Lastly, DHEA has also been shown to exert antiproliferative effects via eNOS activation in ECs [218]. DHEA’s role in these pathways makes it an interesting target for PH, and in fact several studies have investigated DHEA effects in PH with promising results [219–222].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


