Chapter 4 Sedation, Analgesia, and Related Topics
In this chapter the indications, contraindications, and adverse effects of drugs used for sedation and analgesia in the intensive care unit (ICU) are reviewed. In addition, practical tools for the measurement of depth of sedation and quality of analgesia are outlined. The related topics of neuromuscular-blocking drugs and antiemetics are also discussed.
PHARMACOKINETIC CONSIDERATIONS
Duration of Effect
The elimination half-time is the time taken for the amount of drug in the body to decrease by 50%. This parameter is often quoted when describing the pharmacokinetic properties of a drug. However, elimination half-time only rarely reflects the duration of effect. When a drug is given intravenously, it is rapidly distributed to a central “virtual” compartment consisting of plasma, interstitial fluid, and organs with high blood flow (brain, heart, liver, kidneys). This central compartment is in equilibrium with the effect site and with the organs of elimination—the liver and the kidneys. Drugs with low lipid solubility, high ionization, and high protein binding tend to be confined to this central compartment and typically have a small steady-state volume of distribution (VSS; Fig. 4-1). Such drugs can be described using a one-compartment model (V1). For drugs that obey one-compartment kinetics, the duration of effect may be related to the elimination half-time. Examples include aminoglycosides and neuromuscular blocking drugs. However, highly lipid-soluble drugs, including most sedative-hypnotics and opioid analgesics, display multicompartment kinetics in which drugs are redistributed from the central compartment to one or two peripheral compartments (V2, V3; see Fig. 4-1).
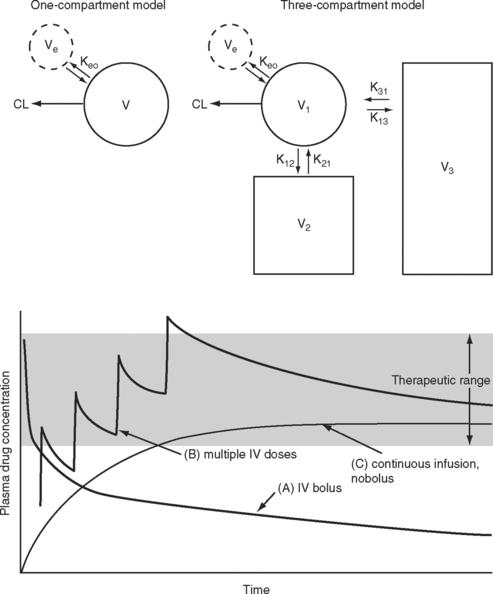
For drugs that display multicompartment kinetics, distribution and context-sensitive half-times are more useful concepts than elimination half-time. The distribution half-time is the time taken for the concentration within the central compartment to fall by 50%. Following a single intravenous dose, the distribution half-time determines the duration of effect of the drug (see Fig. 4-1). The context-sensitive half-time is the time taken for the effect-site concentration to fall by 50% following discontinuation of an intravenous infusion.1 Because a drug accumulates in the peripheral compartments over time, the context-sensitive half-time changes depending on the duration of infusion (Fig. 4-2). The context-sensitive half-time provides some indication of the duration of effect of the drug following both short- and long-term infusions (or repeated bolus doses). The percentage of decrease in concentration required for recovery from a drug’s effect is not necessarily 50%.
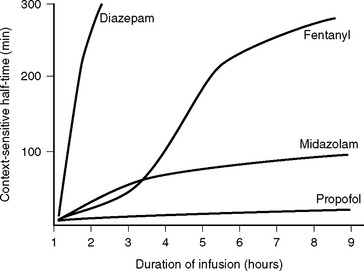
Figure 4.2 Context-sensitive half-times for various sedatives and opioids as a function of time.
(Redrawn from Hughes MA et al: Anesthesiology 76:334, 1992.)
Onset of Action
The speed of onset of a drug depends on multiple factors; two that are of clinical importance for intravenously administered sedatives and analgesics are (1) the speed with which the drug is distributed within the central compartment and (2) the half-time for equilibration between the central and effect-site compartments (T1/2keo; see Fig. 4-1). Low cardiac output slows drug distribution within the central compartment and can greatly prolong the onset time. Thus, when administering potent sedative or analgesic medications to patients with low cardiac output, it is essential to give a small initial dose and wait a longer than normal time for the clinical effect to occur. Values for T1/2keo vary among drugs. For instance, the T1/2keo for morphine, fentanyl, and remifentanil are 17 minutes, 6.6 minutes, and 1.16 minutes, respectively. Therefore, morphine will have a slower onset of action than fentanyl and remifentanil.
Loading and Maintenance Doses
If a drug is administered by constant infusion or repeat doses it takes five (elimination) half-times to achieve 97% of the steady-state concentration (see Fig. 4-1). Thus, for a drug with a relatively long half-time, it is often useful to give a loading dose to rapidly increase the plasma concentration to within the therapeutic range. The loading dose (LD) depends on the volume of distribution (V) of the drug and the desired plasma concentration (CP):
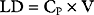
Note that loading dose does not depend on clearance; thus, the loading dose of some renally eliminated drugs, such as gentamicin, do not need to be reduced in patients with renal failure. However, for drugs that display multicompartment kinetics, it is important to be clear which loading dose is used in the calculation: V1 or VSS. A loading dose based on VSS will initially produce very high plasma levels because the drug will be delivered only to the central compartment volume (V1). To avoid this problem, multiple small loading doses based on V1 may be required. This is the approach that is recommended for amiodarone in Chapter 3.
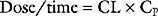
SEDATION
Sedation is part of a continuum of central nervous system (CNS) depression that ranges from anxiolysis through sedation, hypnosis (sleep), unconsciousness, and coma. Most sedative-hypnotic drugs produce anxiolysis at subhypnotic doses. Certain drugs, notably the benzodiazepines, also produce antegrade (i.e., following drug administration) amnesia at low doses. Some sedativehypnotics are anticonvulsants (e.g., benzodiazepines, barbiturates, and propofol). Anxiolysis is not the same as sedation. Antipsychotic drugs produce a state of outward calm but can increase feelings of anxiety and apprehension.
Indications for Sedation
Ventilated patients require sedation to tolerate endotracheal intubation and mechanical ventilation, facilitate nursing care, minimize the stress response, reduce oxygen consumption, diminish recall of unpleasant experiences, and prevent the development of posttraumatic stress disorder.2,3 Less commonly, sedation is indicated in extubated patients for the treatment of anxiety or delirium.
Mechanical ventilation, particularly using lungprotective strategies with long inspiratory times and permissive hypercapnia, is poorly tolerated by nonsedated patients and can result in ventilator dysynchrony (Chapter 29) and the sensation of dyspnea. Distressed patients may become tachycardic and hypertensive, which can exacerbate or provoke myocardial ischemia and bleeding. Such patients may also self-extubate or pull out their intravascular lines and surgical drains. A critically unwell patient commonly benefits from deep sedation, occasionally accompanied by neuromuscular blockade, during the acute phase of an illness. However, most patients do not require paralysis, only a level of sedation sufficient to allow tolerance of endotracheal intubation. Sedation of agitated patients should be commenced only after providing adequate analgesia and treating reversible physiologic causes.3
Adverse Effects of Sedation
Excessive sedation contributes to hypotension and delays awakening, needlessly prolonging the duration of mechanical ventilation.4 Sedation may also mask the development of intracranial, intrathoracic, or intraabdominal complications. The reduction in sympathetic tone that follows the administration of sedative and analgesic drugs can cause important hypotension. Hypotension is particularly marked in patients with high levels of endogenous catecholamines such as those that occur in the settings of hypovolemia and acute heart failure. Following prolonged administration of some sedatives (and opioid analgesics), tolerance may develop such that increased doses are required to elicit the same clinical effect. Abrupt discontinuation of certain sedatives, notably benzodiazepines, in a patient who has developed tolerance, may provoke a withdrawal syndrome (see discussion under subsequent heading Benzodiazepines). For these reasons, the need for sedation should be evaluated on an on-going basis and the depth of sedation regularly assessed.
Assessment of Sedation
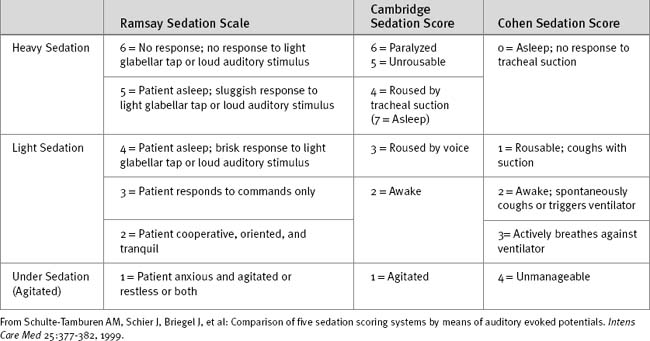
If the clinical state allows, sedation should be stopped each day until the patient shows signs of awakening. Sedation can then be restarted if still indicated. A number of sedation scoring systems have been developed to quantify the depth of sedation and allow sedative drugs to be titrated to effect. Some commonly used scoring systems are shown in Table 4-1. Although primarily a system for monitoring neurologic function after trauma, the Glasgow Coma Scale (Table 4-2) may also be used to monitor sedation, although much information is lost in patients who are intubated and cannot respond verbally. One option in ventilated patients is to revise the Glasgow Coma Scale score to a maximum of 10, with the annotation that the patient is intubated.
| Verbal Response | Motor Response | Eye Response |
|---|---|---|
| 5 = Appropriate | 6 = Obeys commands | 4 = Opens spontaneously |
| 4 = Disorientated | 5 = Localizes to pain | 3 = Opens on command |
| 3 = Unconnected words | 4 = Withdraws from pain | 2 = Opens with pain |
| 2 = Sounds only | 3 = Abnormal flexion to pain | 1 = No response |
| 1 = Nothing | 2 = Extends to pain | |
| 1 = No response | Total = V + M + E = 3-15 |
The Bispectral Index (BIS) monitor, a highly processed electroencephalogram, is widely used to assess depth of anesthesia during surgery and has been used on a limited basis to monitor depth of sedation in the ICU.5,6 A potential problem with this monitor in the ICU environment is that BIS recordings are increased—implying a reduced depth of sedation—by electromyographic activity.7 Thus, BIS recordings tend to be lower in paralyzed patients than in nonparalyzed patients for an equivalent depth of sedation. This phenomenon could potentially lead to paralyzed patients receiving inadequate sedation, resulting in unpleasant awareness.
Sedative Drugs
Propofol
The onset of action following a bolus dose usually occurs within 30 seconds. Propofol has a distribution half-time of 2 to 4 minutes, which results in an offset of effect of 5 to 10 minutes following a bolus dose. There is minimal residual sedation. Propofol has a relatively stable context-sensitive half-time (see Fig. 4-2), and awakening is rapid even after prolonged infusion. In one study of cardiac surgery patients, extubation occurred after a mean time of 7.6 minutes after cessation of propofol infusion (mean dose of 82.8 mg/hr) following 17 hours of continuous sedation.8 The corresponding extubation time for patients given midazolam (mean dose 2.3 mg/hr) was 125 minutes. This rapid offset of clinical effect following prolonged infusion occurs because propofol has high hepatic and extrahepatic clearance (pharmacokinetic effect) and because subhypnotic concentrations of propofol cause minimal sedation (pharmacodynamic effect).
The main side effects of propofol relate to cardiac and respiratory depression. Hypotension due to vasodilation tends to be more marked than with other sedatives. Bolus doses must be used with extreme caution because as little as 20 mg can cause profound hypotension in critically unwell patients. Respiratory depression and apnea are also common, particularly following bolus doses. In extubated patients, equipment for bag-mask ventilation and endotracheal intubation should be immediately available. Doses in excess of 5 mg/kg/hr for prolonged periods have been associated with propofol infusion syndrome. This syndrome is characterized by metabolic acidosis and progressive hemodynamic collapse, and it is potentially fatal.9 Prolonged infusions may result in hyperlipidemia resulting from the intralipid emulsion.
Midazolam
Midazolam can be given enterally or parenterally and has an oral bioavailability of about 50%. For sedation in the ICU, midazolam is given by intermittent intravenous bolus or by continuous infusion. The usual dose range is 2 to 10 mg/hr, but much higher doses are occasionally required. Following a single intravenous dose, midazolam has a rapid onset of action and a short duration of effect. The distribution half-time is about 8 minutes. Bolus doses should be administered slowly (1 mg/min) and titrated to effect because the peak effect may be delayed for several minutes in patients with low cardiac output. Following prolonged infusion the context-sensitive half-time is increased (see Fig. 4-2), which results in a greatly prolonged duration of effect.
Midazolam undergoes hepatic metabolism by hydroxylation—by the cytochrome P-450 (CYP) 3A4 enzyme system—and conjugation. The 1-hydroxy metabolite is pharmacologically active and can contribute to the clinical effect. Drugs that inhibit the CYP3A4 enzyme system (Table 4-3) can prolong the effect of midazolam.
Table 4-3 Selected Substrates, Inhibitors, and Inducers of the CYP3A4 and 2D6 Hepatic Enzyme Systems
| CYP3A4 | ||
|---|---|---|
| Substrates | Inhibitors | Inducers |
| Calcium channel blockers | Antiarrhythmics | Rifamycins |
| Diltiazem | Amiodarone | Rifabutin |
| Felodipine | Calcium channel blockers | Rifampin |
| Verapamil | Diltiazem | Rifapentine |
| Benzodiazepines | Verapamil | Anticonvulsants |
| Midazolam | Nicardipine | Carbamazepine |
| Alprazolam | Azole antifungals | Phenobarbital |
| Immunosuppressives | Itraconazole | Phenytoin |
| Cyclosporine | Ketoconazole | Others |
| Tacrolimus | Voriconazole | St. |
| Sirolimus | Macrolide antibiotics | Anti-HIV agents |
| Statins | Erythromycin | |
| Atorvastatin | Clarithromycin | |
| Lovastatin | Troleandomycin | |
| Macrolide antibiotics | Others | |
| Erythromycin | Grapefruit juice | |
| Clarithromycin | Anti-HIV agents | |
| Others | Metoclopramide | |
| Losartan | ||
| Sildenafil | ||
| Anti-HIV agents | ||
| CYP2D6 | ||
| Substrates | Inhibitors | |
| β blockers | Antidepressants and antipsychotics | |
| Alprenolol | Chlorpromazine | |
| Bufuralol | Haloperidol | |
| Carvedilol | Fluoxetine | |
| Metoprolol | Paroxetine | |
| Propranolol | Clomipramine | |
| Timolol | Doxepin | |
| Antiarrhythmics | Antiarrhythmics | |
| Flecainide | Quinidine | |
| Mexiletine | Amiodarone | |
| Propafenone | Antihistamines | |
| Antipsychotics | H2 antagonists (ranitidine) | |
| Haloperidol | H1 receptor antagonists | |
| Antidepressants | ||
| Fluoxetine | ||
| Paroxetine | ||
| Venlafaxine | ||
| Some tricyclic antidepressants | ||
| Opioids | ||
| Codeine | ||
| Dextromethorphan | ||
| Tramadol | ||
Substrate drugs’metabolisms or inhibitors of the relevant enzyme system. Two are enhanced or inhibited other important CYPenzyme systems are 2C9, which is involved in the metabolism of warfarin, and 2C19, which is involved in the metabolism of the proton pump inhibitors (omeprazole, pantoprazole, etc.)
(Modified from Wilkinson GR: Drug metabolism and variability among patients in drug response. N Engl J Med 352:2211, 2005.)48 HIV, human immunodeficiency virus.
Diazepam
Diazepam may be administered enterally and parenterally and has an oral bioavailability of 100%. Oral or rectal diazepam, in a dose of 5 to 20 mg, is used to treat anxiety and alcohol withdrawal and for night sedation. Intravenously, diazepam is used as boluses of 2.5 to 10 mg as a sedative or anticonvulsant or for acute alcohol withdrawal. Following an intravenous bolus dose, the onset of effect is rapid, similar to that of midazolam. However, the duration of effect is longer because of the relatively long distribution half-time of about 1 hour. Also, the context-sensitive half-time of diazepam is greatly increased following prolonged administration (see Fig. 4-2). Diazepam has several active metabolites that also prolong the clinical effect.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


