Fig. 7.1
An endemic area of schistosomiasis in Northeast of Brazil, where conditions of poor sanitation favor the dissemination of the disease

Fig. 7.2
Biomphalaria glabrata, a snail host of Schistosoma mansoni, found in Brazil
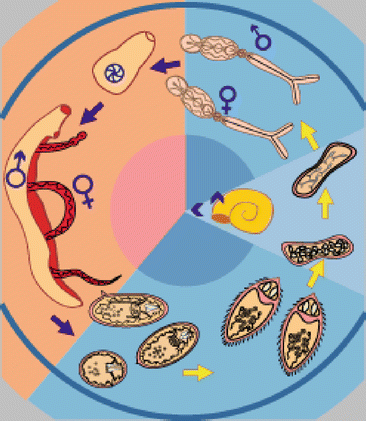
Fig. 7.3
Life cycle of schistosomiasis (WHO 2012): Blue: Eggs from the parasite are released in the water and hatch into miracidia. The miracidia infects the snail host. The snail releases cercariae into water. Orange: Cercariae penetrate the human skin exposed to water and the worms go to target organ where they mate and release eggs. Most of these are excreted in the feces (S. mansoni and S. japonicum) and urine (S. haematobium)
Many eggs that are not eliminated from the body via feces are transported via the hepatic portal vein to the liver where they become lodged and cause granuloma formation. The inflammatory reaction within the granulomas is a stimulus to the production of fibrosis. After many years of infections and reinfections, large sheets of fibrosis can be formed in the liver, depending on the parasite burden and the patient’s immune response leading to a condition known as Symmer’s fibrosis. The hepatocytes are not destroyed, but the fibrosis around the portal vein causes a blockage of hepatic blood flow and secondary portal hypertension. This leads to the opening of portal systemic anastomoses to decompress the portal system [5, 6]. These collaterals may permit the passage of worms and eggs into the pulmonary circulation and induce an immune response and granulomatous reaction in the pulmonary arterial bed leading to pulmonary arterial vascular remodeling and development of PAH in some individuals [7].
Schistosomiasis has a very large spectrum of clinical manifestations [5]. In the acute phase, a macula-papular rash can occur at the site of penetration of cercariae into the skin. This skin reaction occurs some hours after the exposure, can last for several days, and usually disappears spontaneously. It is named cercarial dermatitis and occurs more commonly in people who have not been exposed to the parasite, previously. The lungs can be affected in the acute and chronic phase of the disease. The acute phase, known as Katayama syndrome, occurs predominantly in people that come from non-endemic areas or nonimmune hosts. It is a systemic hypersensitivity reaction that appears a few weeks (16–90 days) after infection and is caused by the passage of migrating schistosomula and egg antigens through the lungs and other parts of the body.
Clinical symptoms include fever, chills, weight loss, headache, anorexia, nausea, vomiting, dry cough, dyspnea, wheezing, and diarrhea. Physical exam may reveal hepatomegaly, splenomegaly, and skin lesions. Liver abscess, brain pseudo-tumors, and myeloradiculopathy are also described in a small number of cases. The symptoms last for 2–12 weeks. Hypereosinophilia is a hallmark of this phase. Eggs of the parasites can be found in the feces about 40 days after infection. A chest X-ray can reveal micronodules, pulmonary condensations, and pleural effusion [2]. On chest computed tomography (CT) nodular lesions can be seen, sometimes with a halo signal and ground glass pattern [8] (Fig. 7.4). This syndrome is believed to be the result of an immunologic process that occurs in nonimmune patients as a reaction to circulating parasite antigens [9, 10].
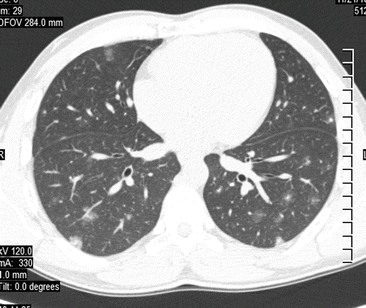
Fig. 7.4
Computed tomography of the thorax of a 29-year-old male patient with Katayama syndrome showing subpleural and peribronchovascular nodules, some with halo sign, and focal ground glass opacities
Chronic schistosomiasis appears several months or years after infection and patients can be asymptomatic or present variable manifestations. The hepatointestinal form can present with abdominal pain and intestinal transit alterations, with or without mild periportal fibrosis and without portal hypertension. This can result in iron-deficiency anemia and constitutional symptoms such as weight loss, malnutrition, and chronic fatigue. The associated hepatomegaly, particularly enlargement of the left lobe with periportal fibrosis, defines the hepatic form. The hepatoesplenic form is associated with splenomegaly in addition to the intestinal symptoms. It is one of the most severe manifestations of the disease, due to portal hypertension that leads to the formation of esophageal and hemorrhoidal variceals that can rupture, leading to severe bleeding that is often the main cause of death in these patients [5, 6].
Patients with chronic S. haematobium infection have chronic inflammation of the venous plexus around the bladder, resulting in hematuria, fibrosis with ureteral obstruction, and nephrolithiasis. Squamous cell carcinoma of the bladder can also develop. Other manifestations of the disease include pseudo-tumor formation, glomerulonephritis, infertility in women, neurologic manifestations, and ectopic disease in the eyes, skin, and urogenital tract [2, 4].
Hepatopulmonary syndrome and pulmonary arterial hypertension (PAH) are the two main types of chronic pulmonary manifestations. Hepatopulmonary syndrome is characterized by the presence of portal hypertension, arterial hypoxemia, and intrapulmonary vascular dilatation [11]. It can be found in 10 % of patients with the hepatosplenic form of schistosomiasis [12]. This form was previously defined as the cyanotic form of schistosomiasis, a more severe phase of hepatopulmonary syndrome, described by Faria et al. [13]. Schistosomiasis-associated pulmonary arterial hypertension (PAH) (Sch-PAH) is defined as an increase in mean pulmonary arterial pressure (mPAP) ≥ 25 mmHg at rest and pulmonary wedge pressure (PWP) ≤15 mmHg in the setting of schistosomotic disease [13].
Epidemiology
As described earlier, nearly 240 million people are infected with the Schistosoma species in the world and about 10 % have severe disease [1, 14]. The true prevalence of Sch-PAH in these individuals is unknown, because the majority of the studies are hospital based with small samples and without a uniform definition of the schistosomal disease and accurate assessment of pulmonary arterial pressure (PAP). A recent systematic review noted that more than 50 % of the studies reporting prevalence of Sch-PAH were from Brazil and less than 20 % from Africa, which accounts for the major burden of schistosomiasis disease [15]. In a rare study that used a random community-based sample, investigators found that the prevalence of Sch-PAH in infected people with schistosomiasis ranged from 0 to 28.3 % (4 ± 8 %). When only people with hepatosplenic disease were considered, the prevalence ranged from 0 to 60 % (18.1 ± 15.9 %) and when the proportion of pulmonary hypertension (PH) was estimated in areas endemic for S. mansoni, it was 6.3–46.5 %. The study concluded that the prevalence of PH is 6.1 % in those infected with schistosomiasis, and 15.1 % in patients with hepatosplenic schistosomiasis, and that Sch-PAH represents 30.8 % of all causes of PH in areas where schistosomiasis is endemic [15]. In these studies, there was no homogeneity in the definition of PH. Some studies defined PH on clinical grounds or histologically. Others assess PH by echocardiogram using different limits of systolic pressures, and others assessed PH by cardiac catheterization. Moreover, several studies did not use exclusion criteria for other causes of PH or other liver diseases in these prevalence studies.
Schistosoma mansoni is the only species present in Brazil where 2.5–6 million people are infected. It continues to cause severe forms of the disease, being responsible for at least 700 hospital admissions and 492 deaths a year. The disease is endemic in nine states and has focal transmission areas in ten states [16]. Two important hemodynamic series found mPAP > 20 mmHg in 23 % of 137 patients [17] and 13 % of 141 patients [18] with portal hypertension. De Cleva et al. [19] found a median mPAP of 20–25 mmHg in 8 (22 %) and mPAP > 25 mmHg in 2 (6 %) of 36 patients with hepatosplenic disease, before they underwent surgery for esophagogastric devascularization and splenectomy or distal splenorenal shunt in order to avoid digestive hemorrhage from esophageal varices. Lapa et al. [20] reported a prevalence of 18.5 % (IC 95 %: 10.8–29.5 %) in patients with a hepatosplenic form of the disease from a PH referral center in Sao Paulo, Brazil. In that study, PH was defined as pulmonary systolic arterial pressure (PSAP) > 40 mmHg as assessed by transthoracic echocardiogram. However, only 7.8 % (IC 95 %: 3.3–16.7 %) had mPAP ≥ 25 mmHg on the right-heart catheterization and only 4.7 % (IC 95 %: 1.5–12.7 %) of all cases had precapillary PH with pulmonary artery occlusion pressure (PAOP) < 15 mmHg. At the same time, PWP has been reported to represent up to 30 % of the PH patients followed up at referral centers in Brazil [21–23].
One limitation of prevalence studies in Sch-PAH is that it is believed that PH occurs almost exclusively in people with the hepatosplenic form of schistosomiasis, which limits the populations studied. These patients represent only 5 % of infected people and this strategy can underestimate the true prevalence [15]. Few studies have included patients without this severe form of schistosomiasis. Lambertucci et al. [24] conducted a study in an endemic area, where 66.3 % of the population had schistosomiasis. Abdominal ultrasound revealed liver fibrosis in 54 % of examined subjects (246) and 25 (11.7 %) of 213 subjects were submitted to echocardiogram showed evidence of PH. However, PASP was estimated by pulmonary arterial acceleration time and the median value found was 31 ± 8.8 mmHg. In another hospital-based study in Brazil, Ferreira et al. [25] found a prevalence of 10.7 % (CI 95 %: 5–19.4 %) using echocardiographic assessment of pulmonary arterial pressure (PAP) in 84 consecutive patients attending an outpatient schistosomiasis clinic who were found to have periportal fibrosis on abdominal ultrasound. The authors included patients with the hepatointestinal and hepatosplenic form of schistosomiasis and defined PH as PASP > 35 mmHg; however, all patients with PH had PASP ≥ 40 mmHg.
Considerably less information is available regarding the prevalence of Sch-PAH in Africa. Studies carried out in the Sudan, Zimbabwe, and Ethiopia found very low levels of Sch-PAH, but these studies were not recent and were not specifically aimed at this problem [15]. In sub-Saharan Africa, where most people infected with schistosoma live, studies of prevalence of PH are lacking.
In general, if we consider that around 200 million people have schistosomiasis in the world, and that of these 120 million have symptomatic disease and 5 % have hepatosplenic disease, approximately 270,000 patients may have Sch-PAH, making it the main cause of PAH in the world [3].
Pathology
Pulmonary vascular remodeling in PAH is characterized by intimal thickening, medial smooth muscle cell hypertrophy, and fibrosis and obstructive lesions of distal precapillary vessels known as plexiform lesions. Inflammatory infiltrate is often demonstrated around the adventitia of the affected vessels. Plexiform lesions consist of a network of vascular channels lined by endothelial cells and a core of myofibroblastic or less well-differentiated cells. These changes are nearly always seen in cases of severe idiopathic PAH (IPAH) and PAH associated with HIV infection, liver cirrhosis, CREST syndrome, and congenital left to right cardiac shunts [26]. These histologic features have also been described in Sch-PAH [27], and, in addition to the similar degree of increase in PAP and pulmonary vascular resistance (PVR), justified the classification of Sch-PAH as group 1 PAH [28].
In the initial pathologic description of Shaw and Ghareeb [29], focal and widespread pulmonary arterial lesions in patients who died of Sch-PAH were attributed to the direct action of the eggs attacking the arterioles. The eggs were demonstrated in the arteriolar lumen producing necrosis of the inner layers of the vessels, with posterior obliteration of the lumen by endarteritis. Intimal thickening, hypertrophy of the media, and collagenous thickening of the adventitia were described along with recanalization by new tortuous vessels called angiomatoid structures. Granulomas were described around extravascular eggs. In 1954, Faria described similar pathologic alterations encountered in specimens of lungs obtained in necropsies: endarteritis in small pulmonary arterial branches, necrotizing arteritis, thrombosis, and intimal fibrous thickenings. However, granulomatous reaction around the vessels was rarely seen and the pulmonary vascular changes found were not directly related to the presence of eggs. The author suggested that the anatomical changes caused by the Schistosoma’s eggs had a secondary role in the pathogenesis of PAH. Interestingly, patients with schistosomiasis without cor pulmonale exhibited eggs retained in smaller branches of the vessels, probably because there was less endarteritis in these cases, permitting the passage of eggs more distally. The most important changes seen were in the intima and media layers of pulmonary arteries and arterioles. The lumen of the vessels could have been filled by concentric or eccentric mass of fibrous tissue associated with medial hypertrophy [30]. Plexiform lesions were demonstrated in more than 80 % of lungs obtained in necropsy studies. These structures continued distally with dilated angiomatoid thin-walled channels [31, 32]. Other authors believed that the embolization of eggs was necessary for the occurrence of arteritis and that PAH would be the result of a peripheral vascular blockage caused by the disseminated granulomata [33, 34].
Recently, a study of 18 lung specimens from individuals who had died from Sch-PAH demonstrated plexiform lesions in all samples and 89 % showed evidence of arterial medial thickening. Only 4 (22 %) of the 18 samples had granuloma, contrasting with prior studies, and S. mansoni eggs in pulmonary samples were not found. A dark pigment was found adjacent to vascular lesions. Its origin is not known, but it had an identical appearance to anthracotic pigment [35].
Pathogenesis
In schistosomiasis, most of the pathology is caused by the T-cell-dependent immune response of the host directed against the eggs of the parasite, which results in chronic granulomatous inflammation and ultimately in fibrosis [36]. The main organ affected is the liver where the eggs of the parasites are trapped along the periportal tissues, which leads to inflammation, tissue eosinophilia, collagen deposition, and fibrosis of the periportal spaces with secondary portal-vein obstruction and subsequent portal hypertension and development of collateral vessels [6]. The granuloma formation is a process which is predominantly CD4+ T helper (Th) cell dependent. The CD4+ Th cell response evolves from a Th1- to a Th2-dominated response following egg production. The type 1 T-cell response occurs with the liberation of several cytokines: IL-1, IL-12, interferon-y (INF-γ), transforming growth factor-β (TGF-β), and TNF-α. They modulate the release of several chemokines. After the liberation of eggs and egg-derived antigens, a Th2 cell response is elicited to limit the primary response, which occurs around 8 weeks from infection. In this phase the liberation of IL-4, IL-5, IL-10, and IL-13 predominates. The formation of granulomas seems to limit the infectious process, in spite of the fact that it provokes the main pathology of the disease [36–38]. The Th1- and Th2-associated cytokines have different roles in the regulation of fibrogenesis. For example, IL-13 has an important role in fibrogenesis whereas TNF-α, INF-γ, and IL-12 seem to have an anti-fibrotic activity and IL-10 has an important regulatory effect in the balance of Th1 and Th2 responses, clearly regulating liver pathology. It is produced from Treg cells and/or Th2 cells. There is some agreement that chronically infected patients with intestinal, hepatointestinal, and hepatosplenic forms of the disease have a dominant Th2 cytokine profile [39, 40], whereas a predominance of the Th1 cytokines is observed in acutely infected patients [36, 37, 39].
Much less is known about the immune mechanisms in Sch-PAH. Today it is recognized that inflammation is an important aspect of most types of PAH and that T-cells, and other immunomodulatory cells are part of this process [41]. In PAH, there appears to be an initial insult to the endothelium, such as hypoxia, mechanical stress, or an infectious process. Secondarily, an inflammatory process occurs which leads to the activation of an immune response and cytokine and chemokine production that propagates this inflammation and leads to the production of growth factors, culminating in vascular remodeling [41]. Genetic determinants may also be important in the development of PAH [42]. In familial PAH, 80 % of patients have a mutation in the gene encoding the bone morphogenetic protein receptor type 2 (BMPR II), a member of the TGF-β superfamily. This mutation is also found in 25 % of patients with IPAH. This mutation is the most important known risk factor to PAH and may interact with environmental agents including chronic infection to initiate the development of PAH. It is not yet known if this type of mutation enhances the development of PAH in patients infected with schistosomiasis.
Perivascular infiltration of inflammatory cells around remodeled vessels has consistently been demonstrated in animal models of PAH, including the mouse model of Sch-PAH and in patients with IPAH [43]. The cells involved are T-cells, macrophages, B cells, mast cells, and dendritic cells in the adventitia and media of muscular pulmonary arteries. With the activation of the immune response, cytokines and chemokines are produced, propagating further inflammation and associated production of growth factors which drive vascular remodeling. In IPAH there is an elevation of serum levels of cytokines, including IL-1-β, IL-6, and IL-8 and chemokines such as chemokine ligand 2/monocyte chemotactic protein-1, CCL5, and CXC3CL1/fractalkine [44]. IL-6 and TGF-β induce differentiation of TH17 cells that are highly proinflammatory. Treg cells also participate in the process balancing Th1 and Th2 responses [41].
In schistosomiasis, the passage of eggs into pulmonary capillaries through portosystemic anastomoses is the main mechanism historically proposed for the development of PAH. PH was felt to occur due to mechanical obstruction of distal pulmonary vessels due to egg impaction, focal arteritis, and inflammation related to the formation of granulomas around the eggs. Crosby et al. [45] showed that infected mice with eggs in the lungs had significant pulmonary hypertension, vascular remodeling, and right ventricular hypertrophy, whereas infected mice without lung egg deposition did not exhibit these changes. Pulmonary vascular remodeling was more severe in arterioles near granulomas, suggesting that the presence of eggs and granulomas drove the process of vascular remodeling in this model of Sch-PAH [45]. However, this model is not entirely applicable to human disease, because Sch-PAH can occur without portal hypertension. Furthermore, in animal models, mice are infected with cercariae and then rechallenged with intravenous administration of eggs that can go directly to the lungs in an attempt to mimic what is supposed to occur in humans via the development of collateral shunts. In this model, infected mice develop a widespread arterial vasculopathy, often with a perivascular inflammatory cellular infiltrate, with pulmonary vascular remodeling affecting media and intimal layers of pulmonary arteries, but without plexiform lesions [35, 43]. These alterations were reduced in mice lacking IL-13RαR1, with loss of the IL-13 function, in a nonstatistically significant manner, while in mice lacking IL-13Rα2, with gain of the IL-13 function, there was an increase in intimal thickness. These infected animals had an increase in right ventricle maximum pressure, mainly if they had a gain of the IL-13 function. Enhanced IL-13 signaling caused PAH in this model. The authors suggested that the enhanced lung inflammatory response triggered by the challenge with eggs correlates with the development of experimental Sch-PAH. The granulomas formed are composed of macrophages, eosinophils, and cells containing smooth muscle actin, which could represent myofibroblasts and/or differentiated smooth muscle cells [43]. Crosby et al. [45] demonstrated that IL-13 but not IL-4 stimulated the migration of pulmonary artery smooth muscle cells in transwell assays. Altered TGF-β signaling was investigated through the expression of Smad2/3 in lungs of schistosomiasis-infected mice and their expression was increased in granulomas and pulmonary arteries. Nevertheless, the absolute levels of TGF-β were not altered in infected mice [45]. In specimens of human lungs obtained at autopsy, there was an intense p-Smad2/3 activity in the smooth muscle cells with thickened media and in vascular channels within the plexiform lesions [35]. Smads are proteins that can be phosphorylated after the attachment of TGF-β in cell receptors and which can move to the nucleus of cells, altering essential cell functions. There is evidence that the TGF-β system stimulates the proliferation of pulmonary arterioles in PAH, and leads to vasculogenesis, including intimal hyperplasia and growth of the medial layer [42, 46]. More recently, Graham et al. [47] demonstrated that a mouse model of Sch-PAH submitted to pharmacological blockage of the TGF-β ligand and receptor and that mice lacking Smad3 were significantly protected from pulmonary vascular remodeling and PAH. This blockage also led to a decrease in IL-4 and IL-13 concentrations [47]. Accordingly, Ferreira et al. [48] demonstrated significantly increased serum levels of TGF-β1 in patients with Sch-PAH compared with patients with schistosomiasis without PAH, suggesting that this growth factor may contribute to vascular remodeling in this disease. Studies using animal models demonstrated a correlation between cytokines and pulmonary remodeling and between the numbers of muscularized small peripheral vessels and the lung egg burden. In these models pulmonary vascular remodeling is characterized by a marked perivascular inflammatory infiltrate, heterogeneous severe thickening of the media of small pulmonary arteries, and the occurrence of plexiform-like lesions in the absence of PAH [45].
It is interesting to note that in the animal model of Sch-PAH, the development of disease requires mice sensitization with cercariae prior to intravenous egg administration, suggesting that a potent pulmonary inflammatory response and inflammatory cell infiltrate are needed rather than the hepatic disease with shunting of eggs and parasites to lungs. The injection of eggs alone was not sufficient to cause right ventricular hypertension, so the embolic disease alone does not seem to cause experimental PAH [43]. Studies with lung samples collected at autopsies of individuals with Sch-PH demonstrated pulmonary vascular remodeling with plexiform lesions and arterial medial thickening in all 18 samples examined, but failed to find eggs of the parasite. Antibodies against S. mansoni-soluble egg antigens (SEA) did not detect significant amounts of egg antigens in the human lung species, although they were identified in lungs of experimentally infected mouse and human intestine specimens. Rare fragments of S. mansoni eggs in human lung specimens were only identified using anti-SEA antibodies within granulomas and not adjacent to the pulmonary vascular lesions. In spite of the presence of vascular remodeling in patients who died of Sch-PAH, significant immunohistochemically antigenic material was not found, suggesting that after an initial acute inflammatory response, vascular lesions are established and can progress or persist independently of the presence of the antigens. These findings corroborate with the persistence of vascular remodeling in patients treated against the parasite [35]. On the other hand, a total or partial regression of liver lesions occurs with parasite treatment [49].
Clinical Presentation of Sch-PAH
In endemic areas, chronic pulmonary disease is common and PH is the most severe complication of the disease. It is almost exclusively found in Schistosomiasis mansoni [7] and is more commonly encountered in patients with hepatosplenic disease. In one study from Brazil, 48.9 % of patients with Sch-PAH had the hepatosplenic form of schistosomiasis, 27.6 % had hepatointestinal disease, and 23.4 % were splenectomized [50]. In Brazil, the splenectomy has been used as a treatment for upper digestive hemorrhage secondary to rupture of esophageal varices [51, 52]. The fact that almost 30 % of patients with Sch-PAH had the hepatointestinal form of the disease suggests that pulmonary embolism of worms or eggs through portosystemic anastomoses is not a prerequisite for the development of PH in schistosomiasis [50].
There are no clinical symptoms that are characteristic of Sch-PAH as compared to other types of PAH. Patients generally have symptoms that are the result of progressive right heart failure such as dyspnea on exertion, weakness, fatigue, cough, giddiness and syncope, palpitation, and chest pain that are the main symptoms [25]. Hemoptysis can also occur. Chest pain is usually caused by right ventricular angina and syncope results from depressed cardiac output and low systemic blood pressure. On physical exam, the lungs are usually clear. Peripheral edema, ascites, anasarca, or jugular vein distension can be seen. A prominent pulmonic component of the second heart sound may be present along with right ventricular heave and murmurs of tricuspid or pulmonic valve insufficiency. Less commonly, patients exhibit cyanosis and rarely digital clubbing is seen. Hepatomegaly secondary to schistosomal disease (left lobe enlargement) or congestion and splenomegaly can be seen in patients with the hepatosplenic form [7, 53].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


