Fig. 13.1
Hematoxylin and eosin stained lung biopsy from a patient with sarcoidosis and severe pulmonary hypertension. (a) A granuloma (black arrow) in the wall of a pulmonary vein (b) A granuloma (black arrow) adjacent to the wall of a pulmonary artery (white arrow) (Images courtesy of Carlyne Cool, MD, National Jewish Health Pathology)
The plexiform arteriopathy that is typically associated with PAH is a known but relatively rare finding in SAPH (see Fig. 13.2) [15]. Interestingly, there is growing recognition that sarcoidosis may increase the risk of thromboembolism [16, 17]. However, it has yet to be validated that this risk is associated with the development of SAPH from either pathophysiologic or epidemiologic studies.
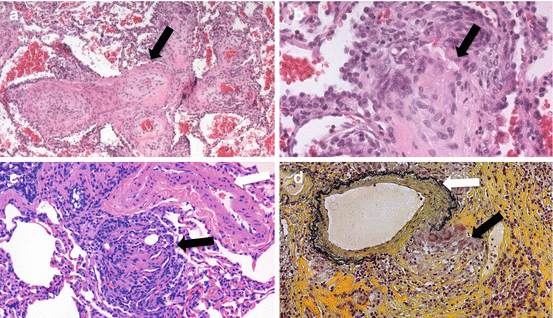
Fig. 13.2
Pulmonary biopsy specimens from: (a) A SAPH patient with plexiform arteriopathy demonstrating marked intimal thickening and hypertrophy (black arrow) (b) A SAPH patient with classic plexiform arteriopathy (black arrow) with intraluminal occlusion by endothelial and modified smooth muscle cells (c) A pulmonary hypertension patient with hematoxylin and eosin-stained biopsy demonstrating a plexiform lesion (black arrow) adjacent to a pulmonary arteriole (white arrow). Note the intravascular plug of endothelial cells forming multiple lamina. (d) A SAPH patient using pentachrome stain (black = elastic tissue, yellow = mature collagen, blue-green = immature collagen, purple = nuclei, red = smooth muscle/fibrin) demonstrating primarily mature collagen, nuclei, and elastic tissue. Note the granuloma i (black arrow) infiltrating the wall of the artery (white arrow) on the lower right (Images courtesy of Carlyne Cool, MD, National Jewish Health Pathology)
Given the potential for comorbid cardiac involvement, SAPH due to left ventricular systolic or diastolic dysfunction remains an important consideration. A pulmonary artery occlusion pressure ≥15 mmHg was found to be present in 29 % in one SAPH series, suggesting left ventricular dysfunction as a contributor to PH [10].
Evaluation
An evaluation for SAPH should be considered whenever dyspnea appears to be disproportionate to the severity of the coexisting pulmonary sarcoidosis, when severe pulmonary sarcoidosis is present, or when right heart failure is present [18]. Echocardiography remains the mainstay of initial non-invasive evaluation for SAPH (see Fig. 13.3). An elevated right ventricular systolic pressure (RVSP) and/or features indicative of right heart remodeling including right ventricular enlargement, hypertrophy, and systolic dysfunction should raise the suspicion SAPH. However, reliance upon the use RVSP as an estimation of pulmonary artery systolic pressure can be misleading, particularly in patients with parenchymal lung disease [19, 20].
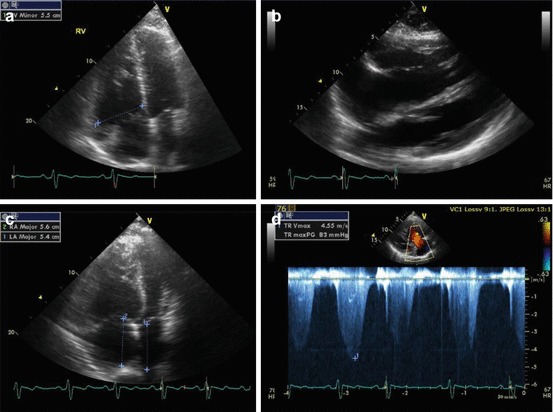
Fig. 13.3
Echocardiographic images from a 47 year old man with SAPH demonstrating significant right ventricular (RV) enlargement in (a) an apical four chamber view (RV major dimension 5.5 cm, normal <4.3 cm) and (b) a subcostal view. (c) An apical four chamber view showing mild right atrial (RA) enlargement (RA major dimension 5.6 cm, normal <5.4 cm) (d) A continuous wave Doppler tracing of tricuspid regurgitation showing a severely elevated right ventricular systolic pressure (83 mmHg, normal ≤35 mmHg)
A multimodality testing approach should be incorporated into the initial SAPH evaluation (see Fig. 13.4). Electrocardiography may indicate the presence of right heart remodeling and corroborate echocardiographic findings. Chest radiographic staging of pulmonary sarcoidosis has utilized the Scadding score and stages patients from I to IV based upon the presence or absence of bilateral hilar lymphadenopathy, pulmonary infiltrates, fibrosis, and bullae [21]. The presence of an advanced Scadding stage should prompt consideration of the SAPH. The finding of ground glass attenuation, septal lines, and extrinsic compression of the pulmonary arteries by mediastinal adenopathy during chest computed tomography has been shown to be associated with SAPH [5]. Main pulmonary enlargement (PA) >29 mm or a ratio of the PA to aortic dimensions >1 has been proposed as a screening tool for PH, but its performance in SAPH has yet to be tested [22]. Surprisingly, neither spirometry, lung volumes, diffusion capacity, exertional hypoxemia, or 6 minute walk distance have proven to be reliably predictive of SAPH [3, 23, 24]. The relatively poor utility of these screening tests likely reflects not only the heterogeneous forms of SAPH but also the inherent limitations of each modality. Therefore, an optimal screening strategy should not rely upon individual test results but rather the composite of multiple tests as well as a symptoms, clinical course, and phenotype before determining whether to pursue diagnostic testing.
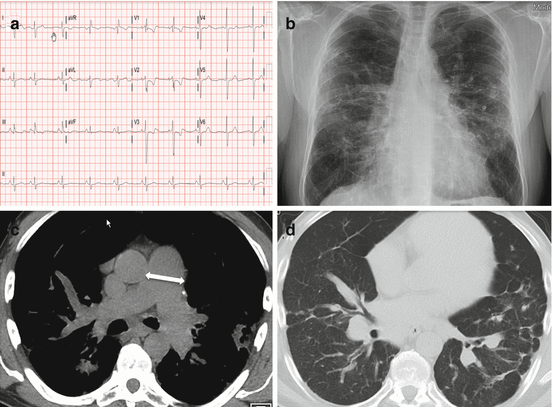
Fig. 13.4
Diagnostic testing from a 47 year old man with SAPH including: (a) an electrocardiogram demonstrating right ventricular hypertrophy and strain as well as right atrial enlargement (b) a chest x-ray showing mid and upper lung parahilar and peripheral reticulonodular opacities consistent with stage IV sarcoidosis (c) a chest computed tomogram (CT) demonstrating pulmonary arterial enlargement (3.9 cm, white arrow) (d) a chest CT showing diffuse lung fibrosis, consolidative opacities, and perihilar nodularity all consistent with pulmonary sarcoidosis
Diagnosis
As with all forms of PH, the diagnosis of SAPH cannot be made noninvasively and requires a right heart catheterization to demonstrate an elevated mean pulmonary arterial pressure (mPAP) ≥25 mmHg [25]. This contrasts the diagnosis of PAH, which also requires elevated mPAP without an elevated left ventricular filling pressure as indicated by pulmonary capillary wedge pressure or left ventricular end diastolic pressure of ≤15 mmHg (see Fig. 13.5).
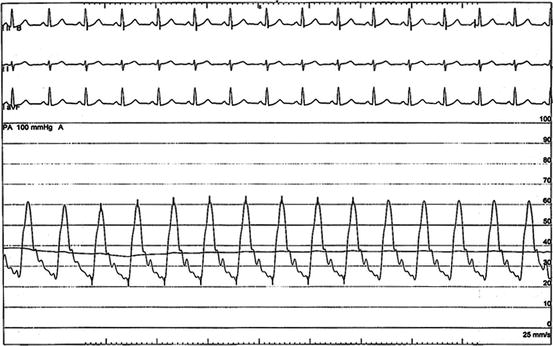
Fig. 13.5
Pulmonary artery pressure waveform from a right heart catheterization in 47 year old man with stage IV pulmonary sarcoidosis and SAPH. The pulmonary artery pressure was 63/23 mmHg (mean 37 mmHg), pulmonary capillary wedge pressure 14 mmHg, and pulmonary vascular resistance of 6 Woods units
Treatment
In contrast to the well-established treatment algorithms for PAH, the optimal treatment approach to SAPH remains an area of controversy and active investigation. However, it is generally agreed upon that modifiable comorbid conditions that potentially contribute to SAPH should be identified and treated, including hypoxemia, obstructive sleep apnea, left ventricular systolic and/or diastolic dysfunction, hypervolemia, and thromboembolic disease. Although corticosteroid therapy is theoretically appealing, its effect on pulmonary hemodynamics in small series has been highly variable and discrepant from improvement in pulmonary mechanics [26–28].
Pulmonary Vasodilators
Although the hemodynamic definition of PH is generally agreed upon, the hemodynamic phenotype that merits the use of targeted pulmonary vasodilator therapy is widely debated. As with other forms of non-WHO classification PH, there is an increasing movement to regard hemodynamically severe or “disproportionate” PH as distinct subtype that may behave like PAH and therefore benefit from drug therapy. When SAPH is viewed as a complication of chronic lung disease, severe PH has been defined as a mPAP ≥35 mmHg or mPAP ≥25 mmHg with low cardiac index (i.e., <2.0 l/min/m2) [29]. Per the 5th World Symposium consensus guidelines, targeted pulmonary vasodilator therapy should be considered on a compassionate basis for this subpopulation with thorough monitoring of gas exchange and inclusion in prospective registries.
Although a growing body of evidence suggests a potential benefit of PAH-specific pharmacologic therapies in SAPH, this literature is limited to small controlled studies, and more commonly, case series. In a recent small, randomized study comparing the endothelin receptor antagonist bosentan to placebo in SAPH, bosentan significantly decreased mean pulmonary artery pressure and pulmonary vascular resistance after 16 weeks of therapy without improving 6 min walk distance [30]. Similarly, in a retrospective study of 12 SAPH patients with end-stage pulmonary sarcoidosis, sildenafil failed to improve 6 min walk distance but did reduce both mPAP and PVR while increasing cardiac index [31]. Prostacyclin therapy has also demonstrated benefit, albeit in very small case series. Use of inhaled iloprost in SAPH improved pulmonary hemodynamics, 6 min walk distance, and quality of life in a subset of patients [32]. However, use of these agents should be tempered by limited data supporting their use as well as concerns that pulmonary vasodilator therapy may precipitate ventilation/perfusion mismatch, particularly in SAPH patients with significant hypoxemia and/or pulmonary fibrosis.
Transplant
The critical role PH plays in the outcomes of sarcoidosis is reflected in the International Society of Heart Lung Transplant consensus guidelines for lung transplantation. Referral for lung transplant evaluation is recommended in patients with New York Heart Association Functional Class III or IV symptom, hypoxemia at rest, right atrial pressure of 15 mmHg, or PH [33].
Prognosis
SAPH patients are more likely to experience debilitating dyspnea, exercise intolerance, and reduced functional capacity [34, 35]. In a population with pulmonary sarcoidosis awaiting lung transplant, the presence of PH predicted a poor prognosis [36, 37]. The estimated 5-year survival for SAPH is only 59 %. Little is known about the impact of pulmonary vasodilator therapy on mortality in SAPH.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


