Fig. 4.1
Proposed mechanism of blood pressure elevation in patients with OSA
Effects of Continuous Positive Airway Pressure (CPAP) on Arterial Hypertension in OSA Patients
In OSA patients, nasal CPAP treatment eliminates repetitive episodes of hypoxia associated with transient cessation of breathing. Short-term nasal CPAP therapy induces a modest decrease in MAP in hypertensive OSA patients [13, 48], and a greater decrease in MAP is seen in patients with higher adherence to CPAP treatment, more severe OSA, and daytime sleepiness [70, 71]. In contrast, in patients with severe OSA without daytime sleepiness, CPAP treatment is found to be ineffective in lowering MAP [8] and reducing the incidence of hypertension or cardiovascular events [7]. Overall, these studies suggest that higher adherence to CPAP treatment is beneficial for patients with severe OSA exhibiting daytime sleepiness.
Autonomic and Cardiovascular Function in Rodents Exposed to IH
Advantages of Experimental Animal Models
Studies in experimental animals involving either rats or mice are very useful, and unlike human studies, they allow modulation of biological systems to elucidate detailed mechanisms of pathophysiology. This experimental approach offers several advantages. First, the effects of IH, a major contributing factor to OSA pathophysiology, can be investigated in the absence of any comorbidities or behavioral alterations normally seen in patients with OSA. Second, autonomic and cardiovascular variables as well as biochemical markers can be measured invasively under well-defined experimental conditions.
Elevation of Blood Pressure in Rodents Exposed to Different IH Patterns
To understand the mechanisms underlying recurrent apnea-induced autonomic and cardiovascular abnormalities, experimental animals exposed to several days of IH have been developed. A survey of the literature showed that investigators in the field have used different patterns of IH. A few of them have been frequently used to examine the consequences and mechanisms of recurrent apneas on autonomic and cardiovascular functions, and these IH patterns are shown in Table 4.1. From a comparative analysis of these studies, the following generalizations can be made: (1) exposure to IH increases blood pressure in rats [37, 63] and mice [90], and (2) the magnitude of the increase in MAP is dependent on the duration of IH, severity of hypoxia, and the duration of hypoxic and normoxic cycles. Nonetheless, these studies suggest that IH in experimental animals induces hypertension. Interestingly, IH-induced increase in blood pressure is absent in carotid body transected rats [37] suggesting that intact peripheral chemoreceptors are necessary for the systemic blood pressure responses elicited by IH.
Table 4.1
Effects of different patterns of intermittent hypoxia on blood pressure (BP) in rats
Investigators | IH paradigm | Effects on BP | BP measurement via |
|---|---|---|---|
Hui et al. [52] | 10 and 21 % oxygen every 90 s during daylight hours | Increased BP after 30 days | Telemetry |
Kanagy et al. [58] | N2-CO2 mixture for 90 s (5 % O2–5 % CO2) followed by 90 s of compressed air to achieve normoxia (21 % O2–0 % CO2) | Increased BP after 7 days | Arterial catheters |
Silva and Schreihofer [106] | 40 s at 6 % O2 every 9 min, 8 h/ day | Increased BP after 2 weeks | Catheters placed into the femoral artery and vein |
Kumar et al. [63] | 15 s of 5 % inspired O2 followed by 5 min of room air (normoxia), 9 episodes per h and 8 h per day for 10 days | Increased BP after 7 days | Tail cuff |
Knight et al. [61] | 10 and 21 % oxygen every 75 s, 80 cycles/day | Significant increase in BP after 7 days | Telemetry |
Zoccal et al. [129] | 5 min of 21 % oxygen and 4 min of pure nitrogen 8 h per day for 35 days | Increased BP after 35 days | Catheters placed into the abdominal aorta |
Fletcher et al. [37] | 3–5 % nadir ambient oxygen every 30 s, 7 h per day for up to 35 days | Increased BP after 35 days | Both tail cuff and femoral arterial catheters |
Joyeux-Faure et al. [56] | 40 s with 5 % O2 and 21 % O2 for 20 s; 8 h during daytime, for 35 days | Increased BP after 5 weeks | Femoral artery |
Effect of IH on Sympathetic Nerve Activity
To assess the role of altered sympathetic tone and levels of vasoconstrictors to IH-induced increase in blood pressure, several studies examine the effects of IH on sympathetic nerve activity and chemo- and baroreflex function and measure the levels of circulating vasoactive hormones in experimental animals. Rats exposed to different patterns of IH showed an increase in cervical [45], renal [51], splanchnic [29], thoracic [131], and lumbar [69] sympathetic nerve activity. In addition, studies by Silva and Schreihofer [106] showed that following IH, the sympathetic nervous system is highly sensitive to stimulation of sciatic nerve and the nasal mucosa. Thus, IH exposure activates sympathetic nervous system similar to that seen in OSA patients.
Effect of IH on Chemo- and Baroreflex Functions
IH has been shown to augment HVR, a hallmark response of arterial chemoreflex in cats [101] and mice [90]. Furthermore, sympathetic response to hypoxia is also exaggerated in rats treated with IH [16, 51, 69], and this effect is abolished by chronic bilateral sectioning of the carotid body [35, 95]. Thus, intact peripheral chemoreceptors are required for the sympathetic responses elicited by IH.
Unlike the arterial chemoreflex, IH-treated adult rats show diminished baroreflex control of heart rate [65] and splanchnic sympathetic nerve activity [87]; however, baroreflex control of cervical nerve activity is unaffected [45]. An increase in baroreflex function is seen in juvenile rats exposed to IH [130] suggesting that chronic IH-induced baroreflex responses are age-dependent.
Effect of IH on Circulating Levels of Vasoactive Hormones
Elevation in circulating catecholamines (both norepinephrine and epinephrine) is also observed in IH-exposed rats [6, 63] and mice [90]. In addition, plasma ET-1 levels are higher in rats treated with IH combined with hypercapnia than in control rats [58], and ET type A receptor antagonist normalized blood pressure in these rats [1].
Effect of IH on Catecholamine Secretion-Synthesis Coupling in the AM
In addition to the sympathetic nerve terminals at the vasculatures, the adrenal medulla (AM) is a major source of circulating catecholamines. To assess whether the AM contributes to IH-induced increase in circulating catecholamines, Fletcher et al. [37] examined the effects of adrenal demedullation on IH-induced raise in plasma catecholamines and blood pressure response. Adrenal demedullation not only prevents the increase in plasma catecholamines but also the elevation in blood pressure in rats exposed to IH, suggesting that catecholamine secretion from the AM plays a critical role in eliciting cardiovascular changes [6].
Given the importance of AM-derived catecholamines in IH-evoked increase in blood pressure, Kumar et al. [63] investigated whether IH alters catecholamine secretion from the AM. Their results show that IH not only increase the content but also augment basal release of norepinephrine and epinephrine from the AM. More importantly, acute hypoxia evokes a robust increase in catecholamine secretion in the AM from rats conditioned with IH, and these responses are absent in control rats. The stimulatory effect of IH on catecholamine secretion is selective to hypoxic stimulus because hypercapnia is found to be ineffective. The hypoxic sensitivity of the AM elicited by IH is associated with downregulation of neurogenically mediated catecholamine secretion as evidenced by reduced responses to nicotine and 2-deoxyglucose [63, 114]. These results suggest that IH evokes functional remodeling of the AM.
The IH-induced increase in catecholamine content in the AM is in part due to increase in enzyme activity of tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine biosynthesis [62]. The increase in TH activity evoked by IH occurs via activation of posttranslational mechanisms involving serine phosphorylation. The activity of TH is physiologically regulated via phosphorylation of one or more of the four serine residues (Ser-8, 19, 31, and 40) located at the N-terminal regulatory domain of the enzyme [31]. IH increases serine phosphorylation of TH at residues 31 and 40, and these changes are associated with upregulation of protein kinase activities [protein kinase A (PKA), extracellular signal-regulated kinases (ERK), and calcium-calmodulin-dependent kinase (CaMK)II] and a simultaneous downregulation of protein phosphatase 2A (PP2A) activities [62, 99]. Thus, IH-induced tilting of the balance between protein kinases and protein phosphatases which leads to sustained phosphorylation and activation of TH resulting in increased catecholamine synthesis (Fig. 4.2) which is necessary to support the increased demand caused by elevated basal and hypoxia-evoked catecholamine secretion (i.e., secretion-synthesis coupling).
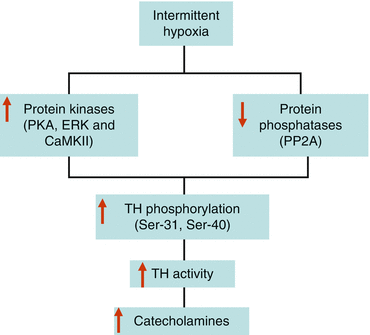
Fig. 4.2
Activation of posttranslational mechanisms in catecholamine synthesis during intermittent hypoxia
Role of Oxidative Stress in OSA-Induced Autonomic and Blood Pressure Changes
The results presented thus far show that adult rats and mice exposed to IH exhibit augmented chemoreflex and depressed baroreflex functions, elevated sympathetic activity, elevated circulating vasoactive hormones, and increased blood pressure similar to those exhibited by OSA patients. How does IH associated with recurrent apneas induce such autonomic and cardiovascular changes? The alternating hypoxia and reoxygenation cycle occurring during IH resembles hypoxia-reperfusion injury, which is shown to induce oxidative stress. Based on this similarity, it has been proposed that reactive oxygen species (ROS) generated during the reoxygenation phase and the ensuing oxidative stress mediate the physiological responses to IH [93].
Oxidative Stress Markers
Various oxidative stress markers have been reported in the literature [116, 119]. ROS oxidize biological molecules forming lipid peroxides, protein carbonyls, and oxidized DNA products and also alter the levels of antioxidants including ascorbic acid, tocopherols, carotenoids, and flavonoids. Changes in the levels of one or more of these oxidation products and antioxidants have been used to assess the occurrence of oxidative stress. Alternatively, the degree of oxidation of biological samples by a known oxidant in vitro can also be used as a measure of oxidative stress. Using these approaches, several studies have examined the occurrence of oxidative stress in OSA patients and experimental animals exposed to IH.
Evidence for Oxidative Stress in OSA Patients
In earlier studies, oxidative stress was assessed by monitoring either free radical-induced formation of diene-conjugate of LDL [121] or hydrogen peroxide-induced lipid peroxidation (by measuring malondialdehyde levels), red blood cell fragility (as an indicator of cell membrane oxidative stress), or total cellular sulfhydryl (thiols) levels [83]. The results from these studies show no significant difference in the levels of these markers of oxidative stress among OSA patients and healthy humans.
In more recent studies, investigators using a larger sample size and a different marker of oxidative stress are able to detect increased oxidative stress in OSA patients compared to control subjects. For instance, Christou et al. [24] report a higher levels of diacron-reactive oxygen metabolites in the blood of OSA patients (n = 21) than in control subjects (n = 5). Likewise, formation of thiobarbituric acid-reactive substance (T-BARS) is higher in patients with severe OSA (59 apneas/h; n = 14) than in healthy subjects (n = 13) [10]. In a larger study involving 114 OSA patients, morning levels of T-BARS and peroxides are found to be significantly higher in OSA patients, with or without cardiovascular disease, than in controls [66, 67]. A recent study by Yamauchi et al. [124] demonstrated that urinary 8-hydroxy-2′-deoxyguanosine excretion was significantly higher in patients with severe OSA (n = 58) compared with control subjects (n = 70). Also, [33] report an increase in the production of ROS in leukocytes isolated from sleep apnea patients compared to healthy subjects.
A reduction in antioxidant capacity also leads to oxidative stress. Analysis of the blood of severe OSA patients (apnea/hypopnea index of >20; n = 14) and healthy humans (n = 5) using the Trolox equivalent antioxidant capacity assay show decreased total antioxidant status in OSA patients [25].
Although results from studies described above indicate the occurrence of oxidative stress in systemic circulation of OSA patients, several other studies report no detectable changes in the levels of lipid peroxidation in OSA patients [2, 83, 117, 121]. These conflicting results may be explained by the small size of the OSA patients and healthy subjects, possible presence of comorbidities and/or medications in OSA patients, the absence of control subjects matched closely for body mass index and obesity, the presence of undiagnosed OSA in control populations, the timing of oxidative stress measurements, and proper handling of the samples to avoid further oxidation [117]. Also, environmental, dietary, and genetic factors can also influence the levels of oxidative stress in humans. It is therefore necessary to include large sample sizes and use highly sensitive and reliable techniques to evaluate accurately the oxidative stress in humans. On the basis of results from large population-based studies, it is safe to conclude that a subset of patients with severe OSA exhibit oxidative stress.
Few studies have investigated the effects of antioxidants on endothelial dysfunction in OSA patients. Either vitamin C infusion [44] or allopurinol treatment [34] seems to improve endothelial dysfunction in patients with severe OSA. Only a limited number of studies have investigated the effect of CPAP on oxidative stress and the results are inconclusive [9, 21, 23, 117, 118].
Oxidative Stress in Animal Models of IH
Evidence for IH-Induced ROS Generation
The possibility of ROS generation in experimental animals during the reoxygenation phase of IH is investigated by several groups. Studies in rats assessed ROS generation by using one or more of the following markers of oxidative stress: levels of T-BARS as index of lipid peroxidation; aconitase activity, which is sensitive to and inhibited by ROS [40]; and isoprostane levels [20]. The results from these studies show that IH facilitates ROS generation in the central and peripheral nervous systems implicated in the control of autonomic and cardiovascular function including the carotid body, the primary peripheral chemoreceptor that detects arterial levels of oxygen [88], carotid sinus region, the site of carotid baroreceptors [87], adrenal medulla, a constituent of the sympathetic nervous system contributing to circulating catecholamines [63, 98, 115], brainstem, regulating sympathetic activity, and cardiovascular function [99, 100, 105]. Also increased ROS generation is observed in rat brain region associated with spatial learning [17, 102] and in left ventricles of rats exposed to chronic IH [22]. Studies in mice also reveal increased ROS generation in brain cortex [122] and brain regions associated with sleep-wake regulation [120]. Unlike IH, sleep deprivation alone has no significant effect on ROS formation in the brain, liver, and skeletal muscle of rats [43]. Therefore, IH, rather than sleep fragmentation, induces oxidative stress in OSA associated with recurrent apneas.
Role of ROS in IH-Induced Responses
To assess the functional significance of increased ROS levels, the effects of antioxidants on autonomic and cardiovascular functions are assessed. Rats are treated daily with manganese (III) tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride (MnTMPyP; 5 mg/kg, IP), a membrane-permeable superoxide dismutase (SOD) mimetic, prior to a 8 h regimen of IH exposure for 10 days. MnTMPyP treatment is found to be highly effective in preventing IH-evoked heightened hypoxic sensitivity, induction of sensory long-term facilitation, and augmented HVR in adults [88, 89] and in neonates [85], as well as depressed baroreflex function [87]. Antioxidant also abolishes the enhanced catecholamine secretion from the AM of IH-treated neonatal [113] and adult rats [63] and mice [64]. Furthermore, antioxidants prevent serine phosphorylation of TH, increases in enzyme activities of TH, protein kinase A, ERK and CaMKII as well as the inhibition of PP2A elicited by IH in the rat brainstem [99]. Collectively, these findings support a critical role for ROS in chemo- and baroreflex and adrenal medullary and brainstem responses to IH.
Furthermore, learning deficits induced by IH is attenuated by the antioxidant PNU-101033E [102]. ROS production and neuronal death associated with IH exposure are absent in transgenic mice overexpressing Cu/Zn-SOD, an antioxidant enzyme [122]. Likewise, pretreatment with green tea catechin ployphenols attenuates IH-induced increase in malondialdehyde levels, an index of ROS generation, as well as neurocognitive deficits associated with IH [17].
Sources of ROS Generation During IH
The IH-evoked ROS elevation could be due to either an increase in ROS synthesis via upregulation of ROS-generating enzymes or a reduction in ROS degradation via downregulation of antioxidant enzymes. NADPH oxidase (Nox) family, xanthine oxidase, and uncoupled eNOS contribute to ROS production, whereas ROS degradation is mediated by antioxidant enzymes that include SOD-1 and 2, catalase, and glutathione peroxidase. ROS is also generated via inhibition of complex I and III of the mitochondrial electron transport chain (ETC; [3]). Regarding the effects of antioxidants on IH, a recent study by Nanduri et al. [76] provides some evidence for IH-induced downregulation of antioxidant enzymes. They report a reduction in SOD-2 mRNA and SOD-2 enzyme activity in the AM of rats exposed to IH. These results suggest that the increased ROS levels caused by IH may involve downregulation of antioxidant enzymes.
Evidence for the Role of Nox
Available evidence suggests that the elevated ROS induced by IH is in part due to increased ROS generation via activation of Nox family of enzymes. For instance, Peng et al. [86] report a 12-fold increase in Nox enzyme activity which is accompanied with an increase in Nox2 mRNA in the carotid body of rats treated with IH. The contribution of Nox2 is supported by studies in Nox2 knockout mice showing absence of IH-induced ROS and functional changes in the carotid body [86]. IH also upregulates Nox2 mRNA and increases Nox activity in the AM of neonatal rats exposed to IH [115]. Interestingly, these authors report that Nox inhibitors prevent acute hypoxia-evoked exaggerated catecholamine secretion as well as increase in [Ca2+]i in the AM of IH-treated neonatal rats. In addition to Nox2, IH also upregulates Nox4 mRNA in the carotid body and AM; however, the functional significance of Nox4 in IH-induced response remains to be investigated. Burckhardt et al. [17] also report an increase in protein levels of p47phox, a Nox subunit, in hippocampal CA1 neuron of rats after 14 days of IH exposure. A similar increase in Nox subunit expression is also reported in sleep-awake brain region of IH-treated mice [120, 128]. Collectively, these studies suggest that Nox is one of the major sources of ROS production in experimental animals treated with IH.
Evidence for the Role of Mitochondrial Electron Transport Chain
In addition to oxidases, mitochondrial ETC is another major source of ROS [3]. Peng et al. [88] show that mitochondrial aconitase activity is lower in carotid bodies from IH-treated rats compared to normoxia exposed controls suggesting generation of ROS in the mitochondria. Studies by Schumacker and his co-workers [47] suggest that continuous hypoxia induces increased ROS production via complex III of the ETC. However, studies in experimental models show that IH selectively inhibits complex I but not III in the rat carotid body [88] and cell cultures [125]. These studies imply that unlike continuous hypoxia, mitochondrial complex I is one of the sources of ROS generation during IH.
Mechanisms of Mitochondrial Complex I Inhibition by IH
How does IH inhibit mitochondrial complex I activity? To test whether Nox-derived ROS mediate mitochondrial complex I inhibition, Khan et al. [60] simultaneously analyzed the activities of Nox and complex I in rat pheochromocytoma (PC)-12 cell cultures exposed to 60 cycles of IH (1.5 % O2 for 30 s followed by 20 % O2 for 5 min at 37 °C). Their results show that (1) IH, in a stimulus-dependent manner, increases Nox activity with a concomitant inhibition of complex I activity; (2) Nox activity returns to baseline values after 3 h of reoxygenation, whereas inhibition of complex I persists even after 24 h of reoxygenation; and (3) either pharmacological inhibition of Nox or siRNA silencing of Nox2 but not Nox4 abolishes IH-induced inhibition of complex I. These results suggest that Nox-derived ROS inhibits complex I in IH-exposed cells. Khan et al. [60] further show that Nox-derived ROS via mobilization of cytosolic Ca2+ to mitochondria facilitate S-glutathionylation of 75- and 50-kDa subunits of complex I. Thus, complex I inhibition by IH occurs via Nox-ROS-Ca2+-dependent posttranslational modification of complex I involving S-glutathionylation reaction. A similar S-glutathionylation reaction is also responsible for the activation of ryanodine receptor-2 which mediates Ca2+ mobilization needed for catecholamine secretion in the AM of neonatal rats exposed to IH [115]. Taken together, these findings provide evidence for functional interaction between Nox2 and mitochondrial complex I which generates a sustained increase in ROS levels via mechanism involving ROS-induced ROS generation (Fig. 4.3).
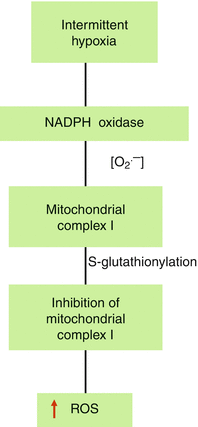
Fig. 4.3
Mechanism of mitochondrial complex I inhibition by intermittent hypoxia
Functional Significance of Mitochondrial ROS
The role of ROS generated by Nox and mitochondria in the elevation of blood pressure by IH is investigated using apocynin (10 mg/kg; IP), a Nox inhibitor and mitochondria-targeted tempol (mito-tempol; 10 mg/kg; IP), which inhibits oxidant formation from mitochondria each day before 8 h regimen of IH exposure for 10 days [60]. Rats treated with vehicle and then exposed to IH for 10 days serve as controls. In conscious rats, post-IH blood pressure is monitored after 1 h (early phase) and 15 h (late phase). In vehicle-treated IH rats, MAP remains elevated both in the early and late phases, whereas these responses are absent in apocynin-treated IH rats. Notably, mito-tempol treatment selectively abolishes IH-evoked elevation in MAP in the late phase without affecting the early phase suggesting mitochondrial-derived ROS contribute to sustained elevation in blood pressure.
Molecular Mechanisms of ROS Generation During IH
Available evidence suggests that transcriptional mechanisms involving hypoxia-inducible factors, HIF-1α and HIF-2α, contribute to sustained oxidative stress elicited by IH. A detailed account of these mechanisms can be found in recent reviews [94, 96]. IH has been shown to increase HIF-1α and decrease HIF-2α protein expression in a ROS-dependent manner [76, 77, 126]. HIF-1α upregulation by IH involves both stabilization and protein synthesis via mTOR pathway [126] and appears to be linked to increased prooxidants (e.g., Nox). The downregulation of HIF-2α induced by IH occurs via calpain-mediated protein degradation and is coupled to transcriptional downregulation of antioxidant enzymes (e.g., SOD-2; [76]). HIF-1α heterozygous mice do not show IH-induced hypertension. On the other hand, blocking IH-induced downregulation of HIF-2α in rats with calpain inhibitor, ALLM, prevents hypertension caused by IH. These findings suggest a novel, hitherto unrecognized roles for HIFs in redox regulation and identify HIFs as one of the major molecular mechanisms underlying IH-induced oxidative stress and hypertension.
Conclusion
In summary, studies in OSA patients and experimental animals exposed to IH show that ROS generated via functional interaction between Nox and mitochondrial ETC contribute to autonomic and cardiovascular abnormalities. These ROS-induced effects seem to involve a complex interplay of redox-sensitive transcription factors, vasoactive hormones/transmitters, membrane receptors, ion channels, and changes in Ca2+-handling (Fig. 4.4). Future studies are needed to define the mechanisms by which IH activates Nox, to delineate potential roles of other cytosolic oxidases in IH-induced ROS generation, and to identify additional cellular targets of ROS under the setting of recurrent apneas.
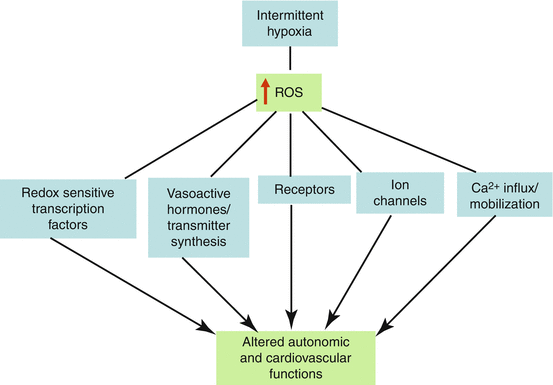
Fig. 4.4
Interaction of ROS with various cellular targets eliciting autonomic and cardiovascular abnormalities during intermittent hypoxia
Acknowledgements
This work is supported by grants from the National Institutes of Health Heart, Lung, and Blood Institute HL-90554, HL-76537, HL-86493, and HL-089616.
References
1.
Allahdadi KJ, Cherng TW, Pai H, Silva AQ, Walker BR, Nelin LD, Kanagy NL. Endothelin type A receptor antagonist normalizes blood pressure in rats exposed to eucapnic intermittent hypoxia. Am J Physiol Heart Circ Physiol. 2008;295:H434–40.PubMedCentralPubMed
2.
Alzoghaibi MA, Bahammam AS. Lipid peroxides, superoxide dismutase and circulating IL-8 and GCP-2 in patients with severe obstructive sleep apnea: a pilot study. Sleep Breath. 2005;9:119–26.PubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


