Fig. 1.1
Pathway and sites of mitochondrial oxidative phosphorylation and ROS formation. Molecular oxygen is activated at complexes I, II, and III to superoxide anion (O2.−) in the mitochondrial inner membrane. Superoxide anion (O2 .−) is dismutated into hydrogen peroxide (H2O2) by the mitochondrial superoxide dismutase (SOD). Iron (Fe) that is redox-active participates in the formation of highly reactive hydroxyl radical (.OH) from H2O2. Thus, the ROS (O2 .−, H2O2, and .OH) generated by mitochondria contributes to oxidative stress which leads to mitochondrial dysfunction, cellular damage, DNA damage, and apoptosis. Antioxidant enzymes such as catalase, glutathione peroxidase (GSH-PX), and thioredoxin (TRX) detoxify H2O2 that is aided by glutathione (GSH) as a cofactor. NADH nicotinamide adenine dinucleotide, FADH 2 flavin adenine dinucleotide, reduced, Q-b cycle ubisemiquinone-cytochrome b cycle
In addition to O2 −. and H2O2, mitochondria is also capable of producing RNS such as NO., generated via breakdown of arginine to citrulline catalyzed by a family of NADH-dependent nitric oxide synthases (NOS). There are at least four isoforms of NOS in mammalian cells: the endothelial constitutive isoform (eNOS) or NOS3, an inducible isoform (iNOS) or NOS2 that is expressed in several cells in response to proinflammatory stimuli and a neuronal isoform (nNOS) or NOS1, and the mitochondrial isoform, mtNOS [21–23]. The existence of mtNOS is still controversial, and recently, another mitochondrial pathway that uses respiratory cytochrome c oxidase as a NO2 reductase to generate NO. has been shown in rats, plants, algae, yeast, mouse brain mitochondria, and human endothelial cells [11]. This pathway, unlike NOS pathway, is oxygen independent and activated by hypoxia or anoxia. While O2 −. is the dominant ROS generated under normoxia, NO. is the major reactive free radical species produced under anoxic conditions, which can combine with O2 −. to form ONOO−. Thus, mitochondria-generated ROS and RNS can regulate mitochondrial activity and function by modulation of mitochondrial fusion, fission, mtDNA damage, lipid peroxidation, and electrical gradient [24–26].
Detoxification of Mitochondrial ROS
ROS production in mammalian cells is highly regulated due to its cytotoxicity. The rates of mitochondrial ROS production and accumulation are balanced through mechanisms that regulate electron transfer in respiratory chain and scavenging of excess ROS. Mammalian cells have evolved a number of antioxidant defense mechanisms such as SOD, catalase, glutathione peroxidase, peroxiredoxins, and thioredoxins to detoxify or scavenge excess mitochondrial and non-mitochondrial ROS.
1.
Superoxide dismutase (SOD): SOD catalyzes the dismutation of O2 −. to H2O2 (Fig. 1.2). There are three isoforms of SOD in mammalian cells including SOD1 (CuZnSOD), SOD2 (MnSOD), and SOD3 (extracellular SOD). Of the three isoforms, SOD2 is exclusively expressed in the mitochondrial matrix [27]. In contrast to SOD1 and SOD2, genetic knockdown of SOD2 causes early neonatal death in mice [28] and endothelial deficiency and dysfunction in apolipoprotein E (ApoE)-deficient mice [29].
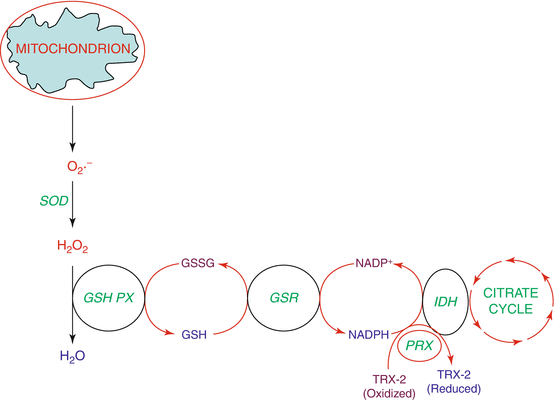
Fig. 1.2
ROS detoxification by antioxidant enzymes and GSH cycling in mitochondria. Superoxide anion (O2 .−) generated by mitochondria is dismutated by mitochondrial superoxide dismutase (SOD) into hydrogen peroxide (H2O2) which in turn is converted into water (H2O) by the mitochondrial glutathione peroxidase (GSH-PX) where in glutathione (GSH) acts a cofactor. GSSG, the oxidized form of GSH, is regenerated back into GSH by the action of GSSG-reductase (GSR) with the participation of NADPH that is generated by isocitrate dehydrogenase (IDH) in the citric acid cycle (tricarboxylic acid cycle). Peroxiredoxin (PRX) regenerates oxidized thioredoxin-2 (TRX-2) to reduced TRX-2 that is also aided by NADPH
2.
Catalase: The enzyme catalase, which is primarily localized in peroxisomes, catalyzes the dismutation of H2O2 to water and O2 (Fig. 1.2). At least three types of catalases have been described, which include the classic Fe heme enzymes, manganese (Mn) enzymes, and the catalase-peroxidases [30]. Although catalase is a key enzyme involved in dismutation of mitochondrial and non-mitochondrial H2O2, knockdown of catalase in mice has no detrimental developmental effects; however, they showed differential sensitivity to oxidant injury. It therefore appears that catalase is dispensable and other detoxifying pathways can compensate [31].
3.
Glutathione peroxidase: Glutathione peroxidase (GPx) reduces lipid hydroperoxides to their corresponding alcohols and further to H2O2 to water (Fig. 1.2). GPx1-4 is a family of isoenzymes homologous to the selenocysteine GPx1 and use reduced glutathione (GSH) as a co-substrate in the reduction of lipid hydroperoxides or H2O2. Not all GPxs use GSH nor do they contain selenocysteine at the active site; however, these are thioredoxin-dependent peroxidases containing a redox-sensitive cysteine or a selenocysteine. GPx1 is the most abundant GPx isoform present in all cells and primarily localized in the cytosol, mitochondria, and in peroxisomes of some cells [32, 33]. In addition to reducing H2O2 and lipid hydroperoxides, GPx1 may also act as a peroxynitrite reductase [34]. However, the in vivo role of GPx1 in modulating peroxynitrite levels is unclear, but lack of GPx1 enhances survival to peroxynitrite [35] through an as yet undefined mechanism. Knockdown of GPx4, but not GPx1, is embryonically lethal [36]; however, GPx1 knockout mice exhibit susceptibility to ischemia-reperfusion injury in mouse myocardium [37]. The effect of knockdown of GPX1 in mice on mitochondrial ROS production is, however, unclear.
4.
Thioredoxin: Thioredoxins (Trxs), with a dithiol/disulfide active site (CGPC), are the major cellular protein disulfide reductases [38], and serve as electron donors for enzymes such as ribonucleotide reductases, thioredoxin peroxidases (peroxiredoxins), and methionine sulfoxide reductases [39]. Trx isoforms are present in most organisms; trx1 localizes within the cytosol and translocates to the nucleus during oxidative stress, whereas trx2 is exclusively located in the mitochondria [40]. Trxs are critical for redox regulation of protein function and signaling via thiol redox control. A growing number of transcription factors including NF-kB or the Ref-1-dependent AP1 require Trx reduction for DNA binding. The cytosolic mammalian Trx, lack of which is embryonically lethal, has numerous functions such as defense against oxidative stress, control of growth, and apoptosis. It is also secreted and has co-cytokine and chemokine activities. Trx reductases of higher eukaryotes are larger (112–130 kDa), selenium-dependent dimeric flavoproteins with a broad substrate specificity that also reduce nondisulfide substrates such as hydroperoxides, vitamin C, or selenite. Mammalian mitochondrial Trx is a monomer of approximately 12 kDa, while mammalian mitochondrial Trx reductase is a selenoprotein homodimer of a 55 kDa subunit. A function of the mitochondrial Trx system is as an enzyme that detoxifies the hydrogen peroxide generated by the mitochondrial metabolism. It is the electron donor for mitochondrial peroxiredoxin. Trx2 deficiency is embryonically lethal at E10.5 d, which coincides with the maturation of mitochondrial function.
5.
Peroxiredoxin: Peroxiredoxins (Prxs) are a family of six small non-seleno thiol-specific peroxidases and are important regulators of cellular ROS. Prx III is localized in the mitochondria, while Prx V is located both in the mitochondria and peroxisomes [41]. All Prxs reduce H2O2 and organic hydroperoxides to H2O and alcohol, respectively, using reducing equivalents from thiol-containing proteins such as Trxs [42]. All the three systems, GPX/GR, Trx/TrxR, and Prx, rely on NADPH as a source of reducing equivalents and mitochondrial NADPH can be regenerated by NADH-supported reduction of NADP+ via energy-dependent trans-hydrogenation. In the mitochondria, the NADPH reduction is carried out by three mitochondrial enzymes: isocitrate dehydrogenase, malic enzyme, and transhydrogenase [43]. As mitochondrial pools of NADPH and GSH are rather large [44, 45], prolonged ROS generation will depend on the antioxidant defense systems that require regeneration of NADPH and GSH. In mice, overexpression of mitochondrial Prx III protected against left ventricular remodeling and myocardial infarction [46]. Thus, mitochondrial redox homeostasis is achieved through cooperation between GSH/GSSG redox coupling in conjunction with redox proteins, GPx, Trx2, and Prx III.
6.
Nuclear factor erythroid 2-related factor 2: Nuclear factor erythroid 2-related factor 2 (Nrf2) is a redox-sensitive transcription factor that is activated in response to oxidative stress and upregulates antioxidant and phase II detoxification enzymes. Nrf2 is activated by oxidizing extracellular conditions that leads to enhanced mitochondrial ROS production, dissociation of Nrf2 from Keap1, and translocation of Nrf2 to the nucleus, binding to the ARE promoter sequences of cytoprotective genes resulting in induction and expression of antioxidant and anti-inflammatory proteins. A potential link between Nrf2, Trx2, and mitochondrial ROS has been recently described [47]. Nrf2 activation can be inhibited by Trx2 overexpression resulting in decreased mitochondrial ROS [48]. Further support comes from decrease in the expression of genes regulated by Nrf2 [49]. Thus, it appears that extracellular redox states mediate Nrf2 signal transduction through ROS generated in the mitochondria and may be regulated by relative concentrations of Trx2. It is noteworthy that other signal transduction pathways, such as TNFα signaling, are also reliant on mitochondrial ROS production, and these studies have shown Nrf2/Trx2 to function as a modulator of signal transduction [50]. Since ROS production is increased as a consequence of oxidizing extracellular conditions and the inhibition of ROS generation decreases Nrf2 activation, it is therefore possible that ROS may be directly responsible for modification of Keap1 enroute to the activation of Nrf2. However, it is currently unknown whether mitochondrial ROS act directly on the Keap1/Nrf2 system or whether mitochondrial ROS affect other regulatory machinery that may indirectly affect signal transduction through (de)activation of other redox-sensitive components. Since oxidant stress conditions are evident in the pathogeneses of many respiratory diseases, furthering our understanding on the mechanistic control of extracellular-induced Nrf2 activation may lead to the development of potential therapeutic interventions. A detailed description of regulation of mitochondrial function by Nrf2 is provided in the accompanying chapter.
7.
Sirtuins: Sirtuins, NAD+-dependent histone/protein deacetylases, catalyze the hydrolysis of acetyl groups from the side chain amino group of lysine residues in proteins. The deacetylation requires NAD+ and generates nicotinamide and 2′-O-acetyl-ADP-ribose [51]. Seven sirtuins members have been identified in mammals, SIRT1–7; SIRT3, 4, and 5 are primarily located in the mitochondria [52]. SIRT1, 6, and 7 are found in the nucleus, whereas SIRT2 is cytosolic, with robust deacetylase activity. There is strong evidence that supports a role for SIRT1 in mounting a response to oxidative stress by directly deacetylating several transcription factors that regulate expression of antioxidant genes. Notably, SIRT1 activates several members of the FOXO family of transcription factors which promote the expression of stress response genes including SOD2 [53–55]. SIRT1 also promotes mitochondrial biogenesis by activating peroxisome proliferator-activated receptor co-activator 1-α (PGC-1α) [56], which increases mitochondrial mass and upregulates the expression of oxidative stress genes including GPx1, catalase, and MnSOD [57]. Also, SIRT1 inactivates the p65 subunit of NF-ĸB through direct deacetylation. NF-ĸB inhibition suppresses the inducible iNOS and nitrous oxide production and thus may lower the cellular ROS load [58]. Mitochondrial SIRT3 deacetylates and activates several enzymes that are critical in maintaining mitochondrial and cellular ROS levels. SIRT3 deacetylates SOD2 at two important lysine residues to boost its catalytic activity, and the deletion of S1RT3 results in loss of catalytic activity of SOD2 [59]. There is evidence that SIRT3 plays an important ameliorative role in preventing the pathological response to pressure overload and aging-associated decline in cardiac function [60]; however, the role of SIRT3 in mitochondrial ROS-dependent respiratory diseases is yet to be defined.
8.
Mitochondria-targeted antioxidant therapies: Given the central role of mitochondria and mitochondrial ROS in human pathologies, several natural antioxidants such as vitamin C, vitamin E, rottlerin, curcumin, ginsenoside Rb1, and epoetin delta have been investigated both in vitro and in vivo for their antioxidant efficacy, and most of these were not found to be effective in attenuating mitochondrial ROS production in response to an environmental stimulus [61]. In the last two decades, there has been considerable advancement in the development of mitochondria-targeted small molecule antioxidants that exhibited reduced mitochondrial oxidative damage and partially prevented decline in mitochondrial function in some of the pathologies. Several of the small molecule antioxidants such as alpha-tocopherol, ubiquinone, piperidine nitroxide, and CP have been conjugated to the lipophilic cation, triphenylphosphonium (TPP), to generate mitochondria-targeted small molecule antioxidants Mito-E2, Mito-Q, Mito-CP, and Mito-TEMPOL, respectively (Fig. 1.3).
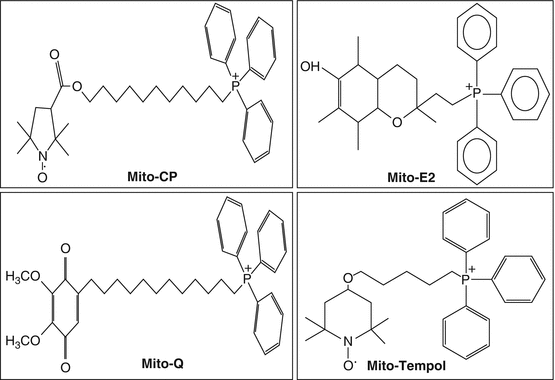
Fig. 1.3
Mitochondrial-targeted antioxidants. Mito-CP mito-carboxy proxyl, Mito-E2 conjugated α-tocopherol moiety to the lipophilic triphenylphosphonium cation (TPP+), Mito-Q conjugated ubiquinol moiety of coenzyme Q to the lipophilic triphenylphosphonium cation (TPP+), Mito-Tempol 2,2,6,6-tetramethyl-4-[5-(triphenylphosphonio) pentoxy]piper-idin-1-oxy bromide
(a)
Mito-Q: Among the four mitochondria-targeted small molecule antioxidants, several studies have focused on protective effects of Mito-Q in animal models of human diseases [62, 63]. Mito-Q administered to rats in their drinking water protected against ischemia-reperfusion in the heart, tissue damage, and mitochondria dysfunction [64, 65]. Moreover, Mito-Q administration prevented organ damage [66] and reduced oxidative stress, IL-6 release, and the levels of biochemical markers of endotoxin-induced cardiac dysfunction [67, 68].
(b)
Mito-CP: In addition to Mito-Q, another TPP-conjugated antioxidant, mitochondria-targeted carboxy proxyl (Mito-CP), has shown promising therapeutic potential against cisplatin-induced nephropathy [69], suppression of medullary thyroid carcinoma cell survival in vitro and in vivo [70], and inhibition of PRx3 and FOXM1 in malignant mesothelioma cell viability [71]. Mito-CP prevented cisplatin-induced mitochondrial injury and dysfunction, renal inflammation, tubular injury, and apoptosis by attenuation of oxidative and nitrative stress mediated by cisplatin [69]. However, Mito-CP stimulated mitochondrial ROS production in medullary thyroid carcinoma cells [70], and malignant mesothelioma cells [71]. These studies suggest that the effect of Mito-CP to scavenge or generate mitochondrial ROS depends on the cell type and dose of Mito-CP used as vitamin C mediates prooxidant effects via Fenton reaction in cells depending upon the balance between concentration and availability of metal ions [72].
(c)
SkQ1: The mitochondria-targeted antioxidant, plastoquinonyl-decyl-triphenylphosphonium (SkQ1), is a conjugate of a lipophilic decyltriphenylphosphonium cation with plastoquinone, quinine, originally discovered in the electron transfer chain of chloroplast [73]. SkQ1, at nanomolar concentrations, exhibits significant antioxidant efficacy both in vitro and in vivo in animal models of ischemia-reperfusion injury in kidney and development of retinopathy [74, 75]. The therapeutic action of SkQ1 on retinopathy in OXYS rats is linked to normalization of gene expression of vascular endothelial growth factor (VEGF) A and pigment epithelium-derived factor (PEDF). The molecular mechanisms of SkQ1-mediated effects are yet to be defined; however, it is clear that SkQ1 is more potent that Mito-Q in increasing survival and health span in animal models of defined pathologies mediated by oxidative stress. The efficacy of SkQ1 that targets on respiratory diseases is largely unknown.
(d)
Mitochondria-targeted cell permeable small peptide antioxidants: A number of small, cell permeable, antioxidant peptides, also known as SS (Szeto-Schiller) peptides, that have a sequence motif that targets them to inner mitochondrial membrane have been reported [76]. These SS peptides (SS-01, SS-02, SS-31, and SS-20) of <10 amino acid residues possess a unique aromatic-cationic sequence motif, which alternates between aromatic and basic residues, enabling them to freely permeate cells in an energy-independent manner [77]. The SS peptides have been studied in vivo and in vitro for protection against oxidative cell death and aging. In neuronal cells, SS-31 offered protection against H2O2-induced cell death [78]. Similarly, both SS-02 and SS-31 protected cultured neuronal and other cell types against environmental toxicants such as 3-nitropropionic acid-, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-, tert-butylhydroperoxide-, and hypochlorous acid-induced mitochondrial dysfunction, ROS generation, and cell death [79, 80]. These cell permeable SS peptides also exhibited beneficial effects in animal models of diseases linked to elevated ROS production and appeared to have least toxic side effects [81, 82]. Additionally, the XJB and related peptides that belong to hemigramicidin-TEMPO compounds were shown to inhibit actinomycin D-induced ROS generation, cardiolipin peroxidation, and apoptosis in mouse embryonic cells [83, 84] and protect cells in culture against radiation damage [85, 86]. The mechanism of action of cell permeable peptides is presumably via antioxidant action, and further studies are required to evaluate the efficacies of these peptides against respiratory diseases.
Mitochondrial ROS in Pulmonary Disorders
Mitochondrion is a key organelle that draws out the latent energy present in substrates such as glucose, fatty acids, and amino acids to generate utilizable energy in the form of ATP. The bioenergetic status in pulmonary disorders is associated with increased ROS generation, antioxidant depletion, and mitochondrial dysfunction leading to organ dysfunction. A better understanding of the association between mitochondrial ROS production, mitochondrial dysfunction, and the severity and outcome of the pulmonary disorder in question is essential for therapeutic strategies targeting mitochondria.
1.
Acute lung injury: Acute lung injury (due to sepsis or ventilator-induced lung injury) and subacute lung injury (due to ionizing radiation-induced lung injury) share profound increases in vascular permeability as a key element driving increased morbidity and mortality. There is evidence demonstrating a potential link between sepsis and mitochondrial dysfunction both in septic patients and animal models of sepsis. Ultrastructural abnormalities in mitochondrial morphology including irregular cristae were reported from liver and skeletal muscle biopsies obtained from septic patients who died in ICU [87]. Similarly, in animal models of sepsis, significant alterations of mitochondrial ultrastructure and abnormalities have been described [88–91]. One postulated mechanism is changes in mitochondrial function due to inhibition of mitochondrial respiratory chain, which may contribute to a decrease in oxygen utilization in acute lung injury [92]. Recent studies have identified the involvement of mediators such as TNF-α, ROS, NO, and peroxynitrite in the inhibition of mitochondrial dysfunction [93]. TNF-α, induced in macrophages and lymphocytes by endotoxin, plays a major role in sepsis-mediated lung inflammation and injury via increase in mitochondrial Ca2+ and ROS production and signaling [94, 95]. In severe sepsis, there is evidence for a massive influx of extracellular Ca2+ via Ca2+ release-activated (CRAC) channel or by opening of voltage-, receptor-, or IP3-operated channels, which is partly the case in mitochondria leading to swelling, mitochondrial dysfunction, and ultimately cell death. While therapies for sepsis are limited, a substantial body of evidence from animal models suggests a beneficial role for mitochondrial targeting with antioxidants such as Mito-Q against endotoxin-mediated sepsis [66]. Further, mitochondria-targeted antioxidants reduced IL-6 release and oxidative stress and improved mitochondrial function in a rat model of acute sepsis [67]. These studies suggest that antioxidants targeted to mitochondria may be an important therapeutic approach in the management of sepsis and multiple organ dysfunction syndrome. A recent study on soluble TNF-α-mediated shedding of the TNF-α receptor 1 ectodomain via increased mitochondrial Ca2+ and ROS and TNF-α-converting enzyme may limit inflammatory response in sepsis [94]. The importance of mitochondrial dysfunction in sepsis was also demonstrated in studies related to administration of bone-marrow-derived stromal cells (BMSCs). Exogenous administration of BMSCs offered protection in mouse models of sepsis-induced acute lung injury [96, 97]. The protection was attributed to Cx43-dependent alveolar attachment and transfer of mitochondria from BMSCs to alveolar epithelial cells [98]. Future studies on mechanisms regulating mitochondrial function may lead to targeted therapeutic approaches for treating sepsis-induced pulmonary inflammation and injury.
2.
Allergic bronchial asthma: Asthma is a complex lung disease characterized by airflow obstruction, airway hyperresponsiveness, and airway inflammation. Exposure to allergens and environmental pollutants induces innate and acquired immune systems to release proinflammatory and bioactive mediators which lead to recruitment and activation of various inflammatory cells such as eosinophils, mast cells, macrophages, neutrophils, lymphocytes, and platelets [99]. Recent studies have revealed the involvement of mitochondria and mitochondrial dysfunction in asthma pathogenesis. Mitochondrial DNA changes and mutations play a role in the development of asthma. Polymorphisms or haplotype differences in the mitochondrial genome may influence the severity of asthma in humans [100, 101]. Human mitochondrial DNA defects have been observed to accumulate with age and in age-related neurodegenerative diseases such as Alzheimer’s and Parkinson’s, which are induced by free radicals and ROS. Although no mitochondrial DNA changes are known to date, age-dependent mitochondrial defects may play a critical role in determining the susceptibility to asthma in the elderly. In addition to mitochondrial DNA, mitochondrial tRNA mutations along with specific rRNA mutations have been significantly more frequent in asthmatic patients compared to control subjects suggesting a role for mitochondrial genetic background in asthma pathogenesis [102]. Involvement of mitochondrial ROS in the pathogenesis of bronchial asthma has received considerable attention in recent years. Several lines of evidence in animal models of asthma and human asthma suggest that ROS released by activated macrophages and granulocytes induces oxidative damage in mitochondria of airway epithelial cells, thereby increasing mucus secretion and epithelial permeability [103–105]. Inhalation of allergens and environmental pollutants induces airway inflammation, releasing proinflammatory mediators such as histamine, PGE2, leukotrienes, and NO. These mediators enhance airway smooth muscle contraction as well as recruit and activate various inflammatory cells into the alveolar space [99], which release ROS, peroxynitrite, and lipid peroxidation products, and they further damage the epithelium. Moreover, the asthmatic epithelium is more susceptible to apoptosis mediated by ROS, probably due to diminished antioxidant levels and decreased endogenous antioxidant enzymes in the peripheral tissues of asthmatic patients [105–107]. Finally, increased numbers of mitochondria and changes in mitochondrial ultrastructure and mitochondrial swelling have been shown in the bronchial epithelium of asthmatic mouse models and human asthmatics [108–110]. Mechanisms underlying increased mitochondrial ROS production are unclear; however, reduced expression of glucocorticoid and estrogen receptors in mitochondria of lung epithelial cells in human asthmatics may be involved in reduction in OXPHOS enzyme biosynthesis, mitochondrial impairment, elevated ROS production, and induction of apoptosis in epithelial and other cell types [10, 111, 112]. Additionally, glucocorticoid and estrogen receptors are involved in regulation of apoptotic, and inflammatory responses [113, 114] and reduced expression of both these receptors could contribute to reduced protection against apoptosis and inflammation in asthma [115]. Interestingly, preexisting mitochondrial dysfunction induced by environmental pollutants intensifies allergic airway inflammation implying that mitochondrial defects could be a risk factor for the development of allergic disorders in susceptible individuals [116]. Although no specific therapy is currently available to minimize mitochondrial dysfunction in experimental models and human asthma, mitochondria-targeted antioxidants may be a promising approach; however, they have yet to be tested as anti-asthmatic drugs.
3.
Pulmonary arterial hypertension: Pulmonary arterial hypertension (PAH) is a lethal disease of the pulmonary vasculature characterized by pulmonary vasoconstriction, right ventricular hypertrophy, and right ventricular failure [117–119]. The hallmark of PAH is excessive proliferation of pulmonary artery smooth muscle cells that thicken the lumen and increase resistance in pulmonary arteries [120]. PAH is a complex vascular disease, and several environmental and genetic factors including hypoxia, loss-of-function mutations of bone morphogenetic protein receptor II, and viral infections have been linked to the development of PAH [121]; however, the cause and mechanisms of the vascular remodeling in PAH remain undefined. Abnormal mitochondria in pulmonary artery smooth muscle cells may suppress mitochondria-dependent apoptosis and contribute to the vascular remodeling in PAH and recent studies indicate a role for Nogo-B in hypoxia-mediated disruption of mitochondria-endoplasmic reticulum stress in mice and human PAH [122]. Several studies have implicated a role for ROS and RNS derived from Nox proteins and mitochondria in the development of PAH [118]. ROS derived from mitochondria may contribute to mitochondrial dysfunction through two pathways. In the first pathway, excess mitochondrial ROS derived from mitochondrial electron transport chain may promote cellular senescence, necrosis, or apoptosis, leading to endothelial dysfunction and vasculopathy [123]. In the second pathway, mitochondrial dysfunction a reduction in H2O2 levels may trigger the pathogenesis of PAH. According to this model, changes in mitochondria result in decreased H2O2 production, leading to decreased cellular redox potential causing cellular depolarization, opening of voltage-gated potassium Kv1.512, influx of Ca2+, and vasoconstriction of pulmonary artery SMCs [124]. Several studies also have indicated impaired NO signaling and bioavailability in patients with PAH and in animal models of PAH [125–127]. Although mitochondrial dysfunction and mitochondrial ROS and RNS are implicated in PAH, potential antioxidant strategies specifically targeting mitochondrial ROS as therapy in PAH have been investigated only in animal models, and they need to be extended to humans.
4.
Pulmonary fibrosis: Pulmonary fibrosis is a lung disease characterized by irreversible destruction of lung architecture, abnormal wound healing, and deposition of extracellular matrix proteins leading to organ dysfunction, disruption of gas exchange, and death from respiratory failure. Lung fibrosis could be idiopathic [128] or arise from exposure to environmental toxins such as fibers, asbestos, metals, pesticides, chemotherapeutic drugs, viruses, multiwalled carbon nanotubes, and radiotherapy [129, 130]. The underlying mechanisms of idiopathic pulmonary fibrosis (IPF) or environmental agents mediated lung fibrosis are poorly understood. However, at the cellular level, injury to the bronchial and alveolar epithelium, epithelial-mesenchymal transition (EMT), activation of macrophages and proliferation, and transformation of fibroblasts to myofibroblasts may all contribute to pulmonary fibrogenesis [131]. At the molecular level, the fibrogenic responses have been linked to growth factors, ROS, TGF-β, matrix metalloproteinases, bioactive lipids such as sphingosine-1-phosphate (S1P), lysophosphatidic acid (LPA), and its G-protein-coupled receptors acting via specific signaling pathways that modulate fibrogenesis in the lung. There is compelling evidence for the role for ROS, TGF-β, TGF-β-mediated ROS, and ROS-dependent TGF-β activation in the development of pulmonary fibrosis in animal models as well as IPF [132–134]. In animal models of asbestos and bleomycin-induced fibrosis and IPF lungs, there is compelling evidence for oxidative stress with increased production of ROS, which could modify macromolecules and alter cellular functions of epithelial cells and fibroblasts [135]. ROS derived from NOX1, NOX2, and NOX4 has been implicated in the pathogenesis of pulmonary fibrosis [136, 137]; however, recent studies with asbestos fibers suggest that activation of mitochondrial ROS and the resultant damage on lung tissue, as observed in a murine model of asbestosis-induced lung fibrosis, are important [138]. Further, macrophage-derived mitochondrial H2O2 seems to play a key role in asbestos fiber-mediated pulmonary fibrosis [138, 139] that was blocked by catalase or knockdown of iron-sulfur protein of complex III in the mitochondrial electron transport chain, a major site of ROS production [140]. Further, accumulating data suggest a potential role for epithelial endoplasmic reticulum stress and mitochondrial ROS in alveolar epithelial cell apoptosis in IPF and asbestos-mediated pulmonary fibrosis [141, 142] (Fig. 1.4). In addition to ROS, dysregulation of TGF-β expression and/or signaling was shown to play an important role in the pathogenesis of pulmonary fibrosis. Interestingly, the interactions between ROS and TGF-β influence the process of fibrogenesis. ROS, derived from NOX and mitochondria, activates latent TGF-β and induces TGF-β expression in many cell types including alveolar epithelial cells and macrophages [143], and conversely, TGF-β also increases ROS production by activating NOX4 [136, 137] and activation of complex III of the mitochondrial electron chain [144]. Mitochondrially targeted antioxidants or genetic disruption of mitochondrial complex III significantly attenuated TGF-β-induced profibrotic gene expression suggesting that targeting mitochondrial ROS may be beneficial in pulmonary fibrosis [144].
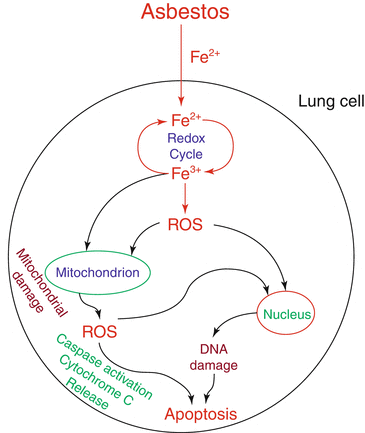
Fig. 1.4
Asbestos-induced ROS generation, mitochondrial damage, and cytotoxicity in lung cells. Asbestos induces cellular ROS generation in lung cells through iron (Fe) redox cycle which cause damage to the mitochondria and nucleus. Asbestos also induces mitochondrial generation of ROS which cause mitochondrial dysfunction and cellular damage including DNA damage, activation of caspases, cytochrome c release, and apoptosis
5.




Bronchopulmonary dysplasia: Bronchopulmonary dysplasia (BPD) is one of the devastating complications of premature newborns with no proven therapy. The development of BPD is associated with a number of risk factors including hyperoxia, ventilator-induced lung injury, and pre- and postnatal infections [145]. An arrest of alveolar development, secondary to mitochondrial bioenergetics failure and reduced ATP production, is one of the hallmarks of BPD [146]. Supplemental oxygen therapy (ventilator) resulting in oxygen toxicity, mediated by production and/or accumulation of ROS, has been implicated in the development of BPD in premature infants and experimental models of hyperoxia-induced injury [147, 148]. Hyperoxia generates excessive ROS, which can directly target and injure alveolar epithelium and lung endothelium; however, the source of ROS in hyperoxia-induced lung injury remains controversial. Some of the earlier reports suggest that mitochondria-derived ROS is a critical determinant of hyperoxic cell damage and apoptosis using respiration-deficient cell lines [149, 150]. Additionally, using nine genetically engineered mice deficient in or overexpression of proteins, a role for mitochondrial oxidants initiating BAX- or BAK-dependent alveolar epithelial cell death contributing to hyperoxia-induced lung injury has been demonstrated [151]. This is in contradiction to an earlier report that showed that hyperoxia-induced cell death to be independent of mitochondrial ROS but requires expression of Bax or Bak proteins [152]. Further, hyperoxia increased ROS formation in pulmonary capillary endothelial cells of rat lungs, which was blocked by complex I inhibitor rotenone as well as inhibitors of NADPH oxidase and the intracellular calcium chelator BAPTA [153]. These data suggest a role for both mitochondrial and NOX-derived ROS from the pulmonary endothelium in situ in hyperoxia-induced lung injury. However, exposure of human lung endothelial cells to hyperoxia enhanced ROS generation that was dependent on NOX activation and independent of mitochondrial electron transport chain [154]. There is no clear explanation for the varying observations on the involvement of mitochondrial vs. NOX proteins in hyperoxia-induced ROS generation; however, use of different cell types, variation due to the use of mouse, rat, and humans, or differential levels of antioxidants in various cell types may all account for the discrepancies. Studies in animal models and premature infants have shown that expression and activity of antioxidant enzymes (AOEs) increase during the third trimester in utero, and premature infants have relative low levels of AOEs [155] that can upset redox balance of the cell leading to apoptosis and inhibition of lung development [151, 156]. Animal studies point out that adult animals are more vulnerable to hyperoxia compared to neonates [157, 158] suggesting reduced cellular antioxidant capacities relative to the newborn animals. Although developmental differences in oxidative stress responses were observed in the murine lung, greater lethality of adult animals exposed to hyperoxia may be due to inflammation rather than to differences in AOEs [159]. The mechanism(s) of increased ROS generation by changes in oxygen pressure from 150 to 300 mm of mercury is unclear, but hyperoxia stimulates signaling pathways mediated by Rac1 [153, 160], MAP kinases [154, 161], PI3 kinase-Akt [162], cytoskeletal redistribution [163], and formation of lipid rafts [164] that contribute to NOX-dependent and NOX-independent ROS formation ultimately leading to lung injury. A recent study suggests the potential involvement of sphingosine-1-phosphate (S1P) and sphingosine kinase (SphK) 1 signaling axis in a murine model of neonatal BPD [165]. Exposure of neonatal mice to hyperoxia enhanced sphingosine-1-phosphate (S1P) levels in lung tissues and Sphk1 −/− , but not Sphk2 −/− or Sgpl1 +/− , mice offered protection against hyperoxia-induced lung injury with improved alveolarization and alveolar integrity compared to their wild-type counterparts. Further, SphK1 deficiency attenuated hyperoxia-induced accumulation of IL-6 in bronchoalveolar lavage fluids and NADPH oxidase (NOX) 2 and NOX 4 protein expression in lung tissue. In vitro experiments using human lung microvascular endothelial cells (HLMVECs) showed that exogenous S1P stimulated intracellular ROS generation, while transfection with SphK1 siRNA or the use of specific SphK inhibitor attenuated hyperoxia-induced S1P generation. Further, knockdown of NOX2 and NOX4, using specific siRNA, reduced both basal and S1P-induced ROS formation. These results suggest an important role for SphK1-mediated S1P signaling regulated ROS in the development of hyperoxia-induced lung injury in a murine neonatal model of BPD [165]. Further studies are necessary to delineate the therapeutic potential of small molecule inhibitors of sphingosine kinase 1 as well as other enzymes involved in S1P metabolism and antioxidant therapy such as thioredoxin treatment [166] on hyperoxia-mediated alveolar simplification, cell death, and mitochondrial dysfunction.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
