Fig. 36.1
Canonical BMP signaling pathways. BMP form a heteromeric complex with type I and type II BMP receptors. Subsequent to this complex formation, the type II receptor phosphorylates the type I receptor, through which the Smad1, Smad5, or Smad8 is phosphorylated. Phosphorylated Smad forms a complex with the common Smad4 and is transported into the nucleus. Besides signaling via Smads, the BMP signal can also be transduced via MAP3K7(Tak1)/MAP3K7IP1 (Tab1), RAS, MAPK1 (ERK), or PI3 kinase. Extracellularly, BMPs can be inhibited by secreted inhibitors, such as NOG, CHRD (chordin), GREM1 (Gremlin), and FST (follistatin), or by the decoy receptor BAMBI, which lacks the intracellular domain for signal propagation. BMP signal transduction is intracellularly inhibited by Smad6 or SMURF (van Wijk et al. [15])
Spatial and Temporal Localization of BMP Signaling in Valve Development
BMPs are uniquely expressed during cushion formation and valve maturation in both the OFT and AV canal (AVC). BMP2 and BMP4 have been widely studied because of their localization within the myocardium along the cushion-forming regions. BMP2 and BMP4 are expressed within the myocardium of the AVC and OFT and thought to be a strong inducers of EMT and cushion formation. In mice, BMP2 expression dissipates in the OFT by E10.5 while continuing to persist throughout the AVC myocardium. On the other hand, BMP4 expression dissipates in the AVC by E10.5 while continuing to persist throughout the OFT myocardium. BMP5 is expressed in the myocardium before and during cardiac cushion EMT (at E8.5) [16]. However, it is later downregulated, and its role in cushion formation or maturation is thought to be minimal. BMP6 is found within the endocardium of the OFT at E8.5 and E9.5 [17], OFT myocardium at E10.5, and in the mesenchyme of the cushions of the AVC at E10.5 [18]. At later stages, however, it has been described in the cushion mesenchyme of the OFT. BMP7 is widely expressed throughout the myocardium with modest expression in AV valve mesenchyme [19, 20].
Traditional mouse knockout models used to define the role of BMP in valve morphogenesis has been challenging. Both BMP2 and BMP4 null mutant mice have early pre-cardiac embryonic lethality [21, 22]. Interestingly, BMP5, BMP6, and BMP7 alone do not produce cardiac defects. However, combinations of BMP knockout mice models have proven to affect valve formation. Within the BMP5/BMP7 double–knockout mouse, cardiac cushions do not form, but the precise role is difficult to determine because of the global disruption in development [16]. BMP6/BMP7 double-knockout mice show a marked delay in the formation of outflow tract cushions because of reduced cell proliferation. Compared with the outflow tract cushions, AV canal cushions are generally less compromised by the loss of BMP6 and BMP7 [17]. This suggests that the combinations of these growth factors are critical for valve morphogenesis.
Analysis of BMP receptor expression patterns and conditional mutations have provided additional insight into the role of BMP in valve formation and remodeling because of their reduced lethality. However, extracting mechanistic understanding is challenging because of their promiscuity. Of the type I receptors, Alk6 (BMPRIB) is not expressed, and Alk3 (BMPRIA) is ubiquitously expressed, while Alk2 (ACVR1) can transduce both BMP and TGFβ signals, is expressed in the heart rather widely [23, 24]. Two type II receptors have been described as being able to transduce a BMP signal. ActRII, originally described as a type II receptor for activins, can also transduce a BMP signal [25], although its pattern of expression in the heart has not been described in detail. The main type II BMP receptor, BMPRII (which mainly binds with Alk3), is ubiquitously expressed in the heart, and all tissues during development [20] (Fig. 36.2).
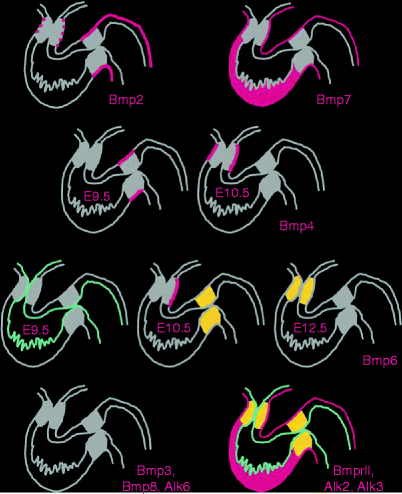
Fig. 36.2
Expression patterns of BMP family genes in the developing mouse heart. BMP2 is expressed only briefly and faintly in the OFT myocardium, but strongly and persistently in the myocardium of the AVC (and atria). BMP4 expression switches from the myocardium of the AVC to the myocardium of the OFT between E9.5 and E10.5. The very dynamic expression of BMP6 is seen first in the endocardium at E9.5, then in the mesenchyme of the AVC cushions and in the myocardium of the left OFT at E10.5, and finally becomes restricted to the mesenchyme of the OFT at E12.5. BMP7 is expressed strongly in all the myocardium at all stages described. BMP3, BMP8, and receptor Alk6 are not expressed in the developing heart, while Alk2, Alk3, and BMPRII are ubiquitous (Delot [20])
Mice lacking type II or type IA BMP receptors die at gastrulation and cannot be used to assess potential later roles in valvular morphogenesis. However, Cre/Lox-targeted deletion of Alk3 in cardiac myocytes revealed an unexpected role for this receptor in the formation of the AVC cushions. EMT in both AVC and OFT regions is normal, suggesting these BMP receptors were either not required or more likely redundant. However, during the remodeling process, cushions were found smaller in size and cushion fusion does not occur. Elimination of Alk3 was also found to diminish TGFβ2 expression in the AVC, which further supports the notion that BMP is an important contributor to the TGFβ paracrine response [26]. Likewise, mice with a hypomorphic BMP receptor II allele (BMPR2) die before birth as a result of cardiovascular abnormalities. Similar to the Alk3 mutant mice, EMT is initiated in both the AV and OFT endocardial cushions. However, OFT cushions failed to develop past the initial EMT events, suggesting that BMPR2 is required for subsequent growth and maintenance of the conotruncal cushions [27].
Growth Factor Regulation of Valve Formation by BMP
Initiation of EMT
Efforts to investigate the mechanisms of EMT and cushion formation have been greatly facilitated by the use of a three-dimensional collagen gel explants. In this system, AV canals (chick or mouse embryos) are isolated and then explanted onto the collagen gel surface. The endocardium of the AV canal adheres to the surface, from which a subset of these cells undergoes the mesenchymal transformation and invades the underlying matrix. Interestingly, EMT does not take place if the myocardium is removed directly after the endocardium has been attached to the collagen gel surface. This suggests that the AV myocardium has a unique capacity to secrete specific signals for EMT [28]. After these landmark observational studies, the focus shifted to understand the makeup of the myocardial inductive signal for EMT in AV canal endocardial cells. Through segmental patterning, it was found that myocardial cells express BMP2 in a manner consistent with the segmental pattern of cushion formation. Using these AV explants in mice, Sugi et al. showed that BMP2 was sufficient to induce EMT in mouse AV canal cultures in the absence of AV myocardium [29]. Furthermore, treatment of AV cultures with Noggin, a BMP inhibitor, prevented mesenchymal cell formation in untreated cultures and also prevented BMP2 induced EMT in AV cultures with intact AV myocardium.
Interestingly, BMP2 treatments of AV explants resulted in increased TGFβ2 protein levels [29]. This is particularly important because antagonists to TGFβ2 and TGFβ3 show that they play distinct but complementary roles in regulating the initial transformation events in chick. Endocardial TGFβ2 expression is required for cell separation and hypertrophy, while TGFβ3 signaling is required for mesenchymal transformation and migration [30]. TGFβ3 induces expression of MMP-2 and MT-MMP, which digest the collagen IV endocardial matrix permitting invasion [31]. At the onset of EMT in chick cardiogenesis, TGFβ3 is expressed in transforming endothelial and invading mesenchymal cells, while BMP2 is expressed in the subjacent myocardium. To restrict this entire process to the cushion forming region in the lumen, endocardial Notch1 signals repress myocardial BMP2 through the Hey1 transcription factor [20]. Furthermore, chick AV explants experiments have investigated the combinatory role of both TGFβ3 and BMP2. In summary, it was found that (1) myocardially derived inductive signals upregulate the expression of AV endothelial TGFβ3 at the onset of EMT, (2) TGFβ3 needs to be expressed by these endothelial cells to trigger the initial phenotypic changes of EMT, and (3) myocardial BMP2 acts synergistically with TGFβ3 in the initiation of EMT [32].
Cushion Maturation
During cushion formation, type I BMP receptors, BMPR1A (Alk3), BMPR1B (Alk6), and Alk2, have all been localized in AV cushion mesenchyme in stage-24 chick embryos. AV explant experiments applying exogenous BMP2 or caBMPR1B (Alk6) treatments significantly promoted expression of an extracellular matrix (ECM) protein periostin, a known valvulogenic matrix maturation mediator whereas periostin expression was repressed by adding Noggin or dnBMPR1B (Alk6)-virus to the culture [33]. Moreover, transcripts of Twist and Id1, were induced by BMP2 but repressed by Noggin in cushion mesenchymal cell cultures. This data provides evidence that BMP2 signaling induces biological processes involved in early AV valvulogenesis, i.e., mesenchymal cell migration and expression of periostin, indicating critical roles for BMP signaling in post-EMT AV cushion tissue maturation and differentiation (Fig. 36.3).
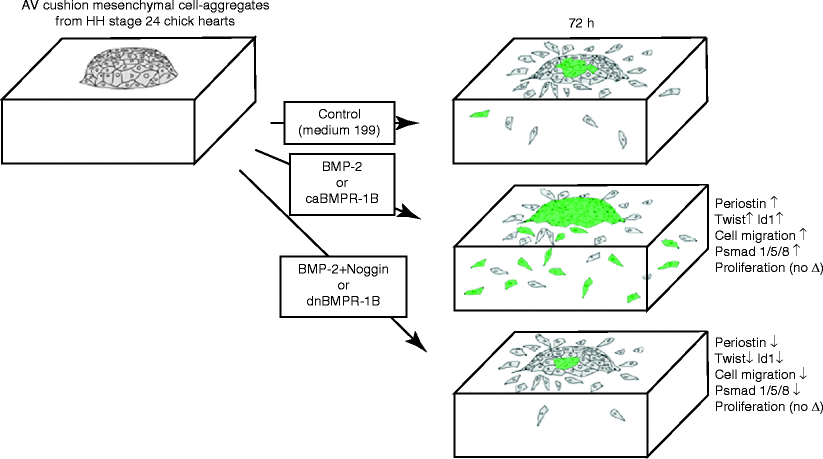
Fig. 36.3
Summary diagram illustrating the results from bioassays to assess the role of BMP2 and BMP signaling using AV cushion mesenchymal cell aggregates cultured on 3D-collagen gels. Exogenous BMP2 or caBMPR1B treatments induced mesenchymal cell migration and expression of periostin, Twist and Id1. In contrast, Noggin or dnBMPR1B treatment inhibited BMP2 promoted cell migration and expression of periostin, Twist, and Id1. Phospho-Smad 1/5/8 expression was induced by BMP2 or caBMPR1B treatments but reduced by Noggin or dnBMPR1B treatments (Inai et al. [33])
BMP2-induced periostin expression plays an important role in the developing cushion and maturation of valves. As a secreted fasciclin domain-ECM protein that associates with areas of fibrosis, periostin can directly interact with other ECM proteins, such as fibronectin, tenascin-C, collagen I, collagen V, and heparin sulfate proteoglycans. Periostin serves as a ligand for select integrins, such as αvβ3, αvβ5, and α4β6, where it can affect the ability of cells (fibroblasts or cancer cells) to migrate and/or undergo mesenchymal transformation in select tissues [34]. Using the aforementioned in vitro assays, Butcher et al. found that upon periostin overexpression, mesenchyme invasion was enhanced in a dose-dependent manner through 3D collagen gels and increased matrix compaction. It was also found that this invasion was dependent on αvβ3 more than β1 integrin signaling and was mediated differentially by Rho kinase and PI3 kinase [35].
BMP4 signaling is thought to be involved in both cardiac cushion EMT and later stage valve remodeling. By combining the use of a hypomorphic BMP4 allele with conditional gene inactivation, BMP4 was found to signal from the myocardium and directly mediate atrioventricular septation. Likewise, more pronounced reductions in myocardial BMP4 expression results in a complete atrioventricular septal defect (AVSD). The AVSDs in these mouse models appear to result from decreased cell proliferation within the AV cardiac cushions [36]. Furthermore, the anterior heart field (AHF)–derived myocardium, another essential source of BMP4, is required for normal endocardial cushion expansion and remodeling. Loss of BMP4 from the AHF in mice results in an insufficient number of cells in the developing OFT endocardial cushions, defective cushion remodeling, ventricular septal defects, persistent truncus arteriosus, and abnormal semilunar valve formation [37].
Downstream signaling through Smad phosphorylation is dependent upon the formation of the Smad1-Smad4 and/or Smad5-Smad4 transcription factor complexes. Smad6 is an important Smad protein capable of attenuating BMP signaling through competition of Smad4 binding [38]. Smad6 is expressed in the AV and OFT regions of the heart during development. Smad6 homozygous knockout mice displayed hypercellular AV and OFT cardiac cushions. The hypercellular cushion phenotype in Smad6 null mice is consistent with BMP mediating either EMT or subsequent mesenchymal cell proliferation within cardiac cushions. In addition, the role of Smad6 in the homeostasis of the adult valves is indicated by the development of bone-related ossification upon Smad6 knockout [39]. It would seem that BMP-induced mesenchyme formation or proliferation is controlled by a negative feedback mechanism involving Smad6 and controlling expression of bone-related genes in adult homeostasis [2].
Transcriptional Regulation of Valve Formation by BMP
The role of BMP directing the differentiation of mesenchymal valve progenitor cells is becoming more understood at the transcriptional level. Recent work by Chakraborty et al. conducted gene expression profiling analysis of murine E12.5 AV endocardial cushions compared with E17.5. They hypothesized the existence of shared regulatory pathways active in developing AV valves and bone progenitor cells. Overall, MC3T3 cells (pre-osteoblasts) were significantly more similar to E17.5 valves than to E12.5 cushions, supporting the hypothesis that valve maturation involves the expression of many genes also expressed in osteoblasts. Several transcription factors characteristic of mesenchymal and osteoblast precursor cells, including Twist1, Sox9, Tbx20, and Msx1/Msx2, are predominant in E12.5 cushion [40]. We will now discuss these transcription factors with regards to valve formation below.
Sox9
Sox9 is thought to be a master regulator required for endocardial cushion cell lineage expansion, as well as differentiation associated with cartilage progenitor cells [41, 42]. In both chick and mouse, Sox9 is expressed within the endocardial cushions and remodeling leaflets. BMP2 activates expression of Sox9 and the cartilage differentiation marker Col2a1, which has been observed in cultured avian endocardial cushion cells [43]. Upon gene knockout of Sox9 in mice, embryonic lethality occurs between E11.5 and E12.5 with hypoplastic endocardial cushions. It is thought that loss of Sox9 inhibits EMT after delamination and initial migration, but before definitive mesenchymal transformation [44]. During targeted loss of Sox9 with Col2a1Cre in the remodeling valve, decreased expression of cartilage-associated proteins Col2a1 occurs. In adult mice, heterozygous loss of Sox9 in Col2a1Cre results in thickened valve leaflets and calcification characteristic of valve disease [42]. This suggests that that a relationship between BMP and Sox9 has a critical role in endocardial cushion formation and valve remodeling.
Msx1/Msx2
Expression of the Msx1 and Msx2 homeobox genes has been shown to be co-coordinately regulated with the BMP2 and BMP4 ligands in a variety of developing tissues. It known that that both Msx1 and Msx2 are crucial downstream effectors of BMP signaling in endocardial cushion. Upon mouse knockout, Msx1 and Msx2 single homozygous mutant mice exhibited normal valve formation, while hypoplastic AV cushions and malformed AV valves were evident in the double Msx1 and Msx2 homozygous mutant mouse. These results support redundant functions for Msx1 and Msx2 during AV valve morphogenesis. In the Msx1/2 mutant embryos, endocardial expression of Notch1, BMP2/4, and NFATc1 is reduced, and patterning of the AVC myocardium is also abnormal, leading to compromised EMT [45]. In addition, loss of both Msx1 and Msx2 were also found to affect secondary heart field and neural crest anomalies related to defects in cell proliferation and migration [46]. Taken together, combined Msx1 and Msx2 mutations lead to a spectrum of cardiac malformations including double outlet right ventricle (DORV), a pulmonary stenosis, atrial and ventricular septal defects, and hypoplastic ventricle [47].
Twist1
Endocardial cushion expression of Twist1 is induced by BMP2 in both chicken and mouse embryos. During valve morphogenesis, Twist1 is expressed throughout the endocardial cushions of the AVC and OFT, and expression is downregulated in the remodeling valves [48]. Shelton et al. performed gain and loss of function studies in avian endocardial cushion cell cultures to demonstrate that Twist1 promotes cell proliferation and migration, while increasing the expression of periostin and MMP-2 [49]. Twist1 activity was examined in transgenic mice with persistent expression in the developing valves. Persistent expression leads to increased valve cell proliferation, increased expression of Tbx20, and increased ECM gene expression, characteristic of early valve progenitors. Among the ECM genes predominant in the endocardial cushions, Col2a1 was identified as a direct transcriptional target of Twist1. Increased expression also leads to dysregulation of fibrillar collagen and periostin expression, as well as enlarged hypercellular valve leaflets prior to birth [50].
Tbx20
Tbx20 is thought to maintain BMP2 expression localized to the AVC region. Tbx20 is strongly expressed in the myocardium of the AVC in both mouse and chick [51, 52]. Mice lacking Tbx20 in the AVC myocardium fail to form the AVC constriction, and EMT is severely perturbed. Furthermore, downstream genes, such as Twist1, Sox9, and Msx1 involved in the EMT initiation were found nearly absent. During re-expression of BMP2 in the AVC myocardium, BMP2 substantially rescues the EMT defects resulting from the lack of Tbx20, suggesting BMP2 is one of the key downstream targets of Tbx20 in AVC development [53]. Furthermore, Tbx20 gain and loss of function studies performed in chicken AVC explants were found to increase cell proliferation and migration while repressing ECM maturation. Tbx20 promotes expression of the ECM remodeling enzymes, MMP9 and MMP13, while repressing expression of the chondroitin sulfate proteoglycans, aggrecan, and versican [54]. Overall, Tbx20 has essential roles in regulating AVC development that coordinate early cushion formation.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


