Fig. 7.1
Sudden death rates in the general population, post-myocardial infarction (MI) and in patients with reduced ejection fraction [6]
7.3 Mechanisms of Sudden Death in Heart Failure
Ventricular arrhythmias arise from a complex environment that involves adverse remodeling of myocardial electrophysiology coupled with waxing and waning of neural and hormonal signals that arise in response to altered strain on the damaged myocardium (Chaps. 46 and 52). Abnormalities in renal function due to poor perfusion in patients with heart failure may also contribute directly to progression in an arrhythmogenic substrate. Broader understanding of the link between the pathologic changes leading to progressive pump failure and adverse electrical remodeling will open new options for therapeutic intervention.
Cardiac electrophysiologic adverse remodeling is the consequence of structural heart disease, many times resulting from myocardial ischemia or infarction [31–37] (Fig. 7.2), but is clearly seen in patients with dilated cardiomyopathy or hypertrophic cardiomyopathy [38] (Chap. 16). The scope of this chapter is to review the primary mechanisms and therapeutic opportunities to prevent sudden death in patients with heart failure, but it is important to consider that sudden death can occur at any point in the progression from normal to the end stage of heart disease associated with clinical heart failure (Fig. 7.1).
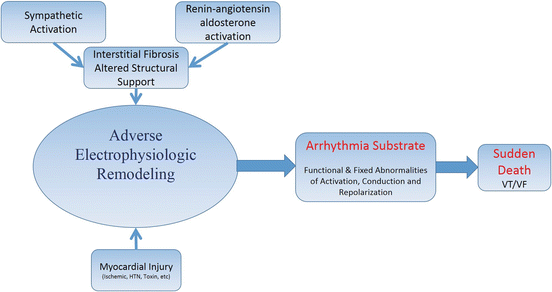
Fig. 7.2
Progression of the arrhythmogenic substrate responsible for the development of ventricular tachycardia (VT) and ventricular fibrillation (VF) associated with sudden cardiac death in heart failure patients
Animal models have provided unique insight to this progression from normal to end-stage heart failure in the same subjects. Certain pathophysiologic themes emerge during this progression which point to the importance of autonomic responses to injury and associations between myocardial substrates conducive for reentrant or triggered arrhythmias. As this pathophysiology progresses, so does the risk for lethal arrhythmias. Even in animal models of sudden death, innate abnormalities of cardiac autonomic control can be seen prior to the onset of cardiovascular disease. Specifically, animals which develop ventricular fibrillation (VF) in response to coronary artery occlusion at the time of myocardial infarction (MI) are characterized by weak vagal reflexes and evidence for elevated sympathetic control [39–42]. Myocardial damage worsens this imbalance, which is associated with the development of specific alterations in regional activation and heterogeneity of repolarization. Individuals at high sudden death risk seem to progress rapidly to “arrhythmogenic heart failure” as left ventricular dysfunction develops due to progressive ischemic heart disease [42]. Neural and hormonal responses to left ventricular dysfunction result in multiple abnormalities that increase the likelihood of lethal arrhythmia development.
Clinical corroboration of the concepts observed in animal modeling is suggested by the findings that patients who died suddenly without prodromal symptoms, otherwise “normal,” usually have autopsy evidence for significant obstructive coronary artery disease usually with a very recent thrombotic event (35–37). This would suggest that the first “symptom” of acute coronary occlusion in these individuals is a lethal arrhythmia. Further evidence is provided by the few patients who survive unexpected cardiac arrest without a prior history of cardiovascular disease, which characterizes them with significant cardiac autonomic control system dysfunction resulting in imbalances favoring strong sympathetic input with relatively weak vagal control [43].
Following myocardial infarction, autonomic imbalances favoring the sympathetic nervous system coupled with depressed vagal reflex activation to a blood pressure increase, i.e., a depressed “baroreflex sensitivity test,” are specifically effective in identifying high-risk experimental animals and in patients with left ventricular dysfunction [39–42]. Relevant to this clinical information is the fact that, experimentally, vagal augmentation by electrical stimulation of the cervical sympathovagal trunk significantly reduces the incidence of ventricular fibrillation when applied to high-risk animals [44]. In contrast, vagal inhibition by atropine [45] or sympathetic activation by ephedrine [46] also enhances arrhythmic risk and mortality in otherwise low-risk subjects. Patients with conditions such as myocardial infarction or heart failure, which are characterized by an autonomic milieu consisting of parasympathetic withdrawal, or inherently weak cardiac vagal control, coupled with sympathetic activation, are at increased relative risk for lethal arrhythmias [47, 48]. Thus, patients have an increasing risk for sudden death as their heart disease progresses from coronary occlusion to heart failure (Fig. 7.1). In this regard, patients with heart failure represent one of the highest-risk groups that are routinely under physician’s care (Chaps. 5 and 6).
7.3.1 Mechanisms of Ventricular Arrhythmias in Heart Failure: Electrophysiologic Adverse Remodeling
Cardiac electrophysiology in heart failure patients is the result of complex interactions between local events, such as myocardial ischemia, fibrosis, and replacement of the normal myocardial syncytium coupled with functional modifications by cardiac control systems primarily arising from the autonomic nervous and renin-angiotensin-aldosterone systems (Chap. 35). Adverse tissue and architectural remodeling, characteristic of chronic heart failure, is not homogeneous, which has direct consequences on cardiac electrical properties. In addition to cardiac structural changes, remodeling of cardiac ion channel densities, structure, or function, coupled with alterations in cell-to-cell communication, significantly alters both activation and repolarization. This process, called “electrical remodeling,” represents the upstream process by which an arrhythmogenic substrate provides risk for lethal tachyarrhythmias (Fig. 7.2) [49–54] (Chap. 47).
7.3.2 Alterations in Activation and Risk for Sudden Death
Cardiac cell activation and conduction of the action potential through the myocardial syncytium depend on the availability of appropriate sodium current and cell-to-cell communication through the proper density and distribution of gap junctions [55–58]. Adverse remodeling associated with chronic cardiovascular disease and heart failure can alter activation and conduction by influencing sodium channel (INa) densities especially in areas of myocardial ischemia or infarction. INa function is also altered in heart failure by changes in cytoskeletal support of the channel [53, 58–63]. Conduction abnormalities can arise from altered INa or by functional abnormalities imposed by intracellular fibrosis or changes in cell size.
These aspects of electrical remodeling can result in a “fixed” arrhythmogenic substrate when changes in the ventricular myocardium permanently alter electrophysiologic characteristics in the area of change. Examples of fixed electrophysiologic abnormalities include fractionated activation across a “mottled” infarct created by surviving tissue interspersed among necrotic or fibrotic tissue [64–70]. Activation fronts encountering areas of patchy fibrosis or fatty necrosis either alter conduction impedance or allow conduction through the zone by way of surviving tissue that forms conduction channels that traverse the area of patchy necrosis. Impulses conducting through channels can exit from the zone of patchy conduction to encounter excitable tissue and give rise to triggers for reentrant arrhythmias. This combination of delayed activation, unidirectional block, and discontinuous conduction is thought to be the macroscopic mechanisms responsible for reentrant tachyarrhythmias.
Additional influences on activation electrophysiology include changes in anisotropy or cell-to-cell communication arising from altered gap junctions producing delayed conduction in the affected areas. Myocardial ischemia or left ventricular hypertrophy results in the downregulation of connexin 43 (Cx43), as well as a redistribution of this protein to the lateral aspects of the myocyte [71–74]. The stimuli for gap junction remodeling are not completely understood, but likely rely on neural and hormonal control of expression and distribution kinetics.
Activation alterations produce fractionated wave fronts that are detectable in heart failure patients at high sudden death risk [68, 69]. The mechanisms involved in establishing fixed changes in activation involve stimulation of fibrosis and fatty necrosis by angiotensin II, aldosterone, and ß-adrenergic receptor activation (Table 7.1). Therefore, neurohormonal intervention is a critical means to prevent permanent changes in the myocardium that produce a fixed substrate for arrhythmias. It is unclear if continued neurohormonal antagonism results in reversal of the electrophysiologic substrate for lethal arrhythmias once that substrate is established. What is clear, however, is that altered activation and conduction, especially when coupled with repolarization heterogeneity, are key components of the high-risk substrate [75, 76].
Table 7.1
Hypothesized effect of neurohormonal intervention to prevent the development of an arrhythmogenic substrate or suppress arrhythmia triggers in cardiovascular disease (see text for detailed references)
Drug class | Action | Antiarrhythmic effect | SCD reduction |
|---|---|---|---|
ACE inhibitors | Antifibrotic | Prevent fixed substrate | Minimal effect in chronic heart failure |
Aldosterone antagonism | Antifibrotic | Prevent fixed substrate | ↓ 34 % heart failure |
Beta-blockade | Antiadrenergic, antifibrotic, reduction in hypertrophy, antiapoptotic | Prevent fixed substrate and suppress functional triggers | ↓ 41 % heart failure |
Bisoprolol | |||
Metoprolol succinate | |||
Carvedilol |
7.3.3 Alterations in Repolarization and Risk for Sudden Death
Prolonged repolarization is characteristic of ischemic heart disease and heart failure pathophysiology resulting in lengthening of the myocardial monophasic action potential [77–92]. Typically, changes in ventricular function, tissue, and electrical remodeling in heart failure are heterogeneous, which is especially true for repolarization abnormalities [78–89]. Repolarization heterogeneity can produce severe repolarization dispersion which is found in subjects at high risk for lethal arrhythmias [78]. Interestingly, lower-risk individuals seem to have much less repolarization heterogeneity compared to their high-risk counterparts. Why some individuals develop severe repolarization heterogeneity is complex, but likely relates to control system responses to injury, such as the degree of sympathetic activation following myocardial infarction. Abnormal repolarization can be detected in humans and animals at high sudden death risk in a variety of ways, including microvolt T-wave alternans [79, 80], increased QT interval dispersion, or heterogeneous ventricular repolarization by high-density mapping [78].
Prolongation of action potential duration may have an adaptive purpose. When the repolarization phase of cardiac tissue prolongs, it increases the likelihood that a reentrant wave front may encounter refractory tissue, which would serve to terminate the circuit. With this effect, repolarization prolongation has the potential to be antiarrhythmic, but the prolongation must be global and homogeneous for the effect to prevent an unstable ventricular electrophysiologic environment. These conditions are seldom encountered when adverse ventricular remodeling occurs.
Prolonged repolarization in heart failure arises because of changes in expression of functional ion channels responsible for the plateau and rapid repolarization of the monophasic action potential [83–93]. Repolarization abnormalities that contribute to the arrhythmogenic substrate arise from both acute changes in autonomic control [90] and from ion channel remodeling in the presence of left ventricular hypertrophy or stretch. Most consistently, the downregulation of the transient outward potassium channel, Ito, which contributes to the plateau phase of the monophasic action potential, is encountered in cells from most etiologies of heart failure. Other potassium channel abnormalities that occur in heart failure include reduced delayed rectifier (IK) current density, coupled with faster deactivation kinetics and slow activation properties, which result in prolongation of the rapid repolarization phase of the monophasic action potential. Left ventricular hypertrophy results in a significant downregulation of the inward rectifying potassium channel, IKs, which becomes the predominant channel responsible for myocyte repolarization in states of sympathetic activation. Along with changes associated with hypertrophy, cytoskeletal support protein remodeling also influences potassium channel function [93]. When regional tissue remodeling is present, with areas of compensatory hypertrophy, the resulting heterogeneous repolarization kinetics increase the risk for spontaneous ventricular tachyarrhythmias (Chaps. 46 and 47).
Global prolongation of ventricular repolarization in patients with heart failure arises from multiple mechanisms [76], which include potassium channel dysfunction, as well as abnormalities in calcium homeostasis [94–100]. It must be recognized that calcium handling is a key component that gives rise to decreased force of contraction, but abnormalities in calcium kinetics also have direct electrophysiologic ramifications [94]. Cells from failing ventricles exhibit a decrease in calcium transient amplitude and its rate of decay. Heterogeneity of calcium transient amplitude and delay when comparing endocardial, mid-myocardial, and epicardial regions, as well as global changes associated with regional heterogeneity and ischemia in the failing heart, very likely contribute to ventricular electrophysiologic instability [96]. Further alterations in the sarcoplasmic reticulum Ca2+-ATPase (SERCA2a), phospholamban, and the sodium-calcium exchanger proteins are described in the failing ventricle and can alter the intracellular calcium concentration [97]. This is associated with ryanodine receptor defects, which when combined with other alterations in calcium homeostasis increases the risk for calcium-dependent afterdepolarizations (Chap. 4). Afterdepolarizations are key elements that form the basis for triggered arrhythmias and sustained polymorphic ventricular tachycardia.
Heterogeneous ventricular repolarization kinetics that result in regional and global prolongation are induced by both acute and chronic changes in neural and hormonal control of the heart. Therefore, substrate changes resulting in fixed repolarization abnormalities can potentially be prevented by neurohormonal intervention, such as aldosterone antagonism or beta-blockade, when applied early in the adverse remodeling process. Chronic neurohormonal intervention also provides benefit by preventing functional or momentary abnormalities in repolarization induced by acute neural activation in response to further injury or physiologic needs.
7.3.4 Hormonal Activation and Sudden Death Risk
The development of permanent (fixed) electrophysiologic substrates can be prevented by early use of neurohormonal interventions including angiotensin-converting enzyme (ACE) inhibitors [101], aldosterone antagonism [102], or antiadrenergic intervention with beta-blockers [103–108] (Table 7.1). Although the direct mechanisms involved in neurohormonal induced electrical remodeling are not fully characterized, general conclusions can be drawn from clinical trial observations. For example, ACE inhibitor therapy reduces mortality primarily by preventing pump failure deaths presumably by inhibiting progressive architectural changes leading to inefficient left ventricular function [109–115]. However, if applied following myocardial infarction (without the requirement for heart failure), ACE inhibitors seem to reduce the incidence of sudden death by as much as 20 % demonstrated by meta-analysis techniques [101].
One can speculate that ACE inhibitor intervention may be effective in reducing sudden death early after myocardial injury by inhibiting angiotensin II-induced interstitial fibrosis leading to fractionated activation characteristic of arrhythmogenic substrates [112, 116]. Aldosterone stimulates proliferation of cardiac fibroblasts resulting in increased interstitial collagen formation which may influence arrhythmogenic substrate development over time. Other effects of angiotensin-aldosterone activation, such as promotion of inflammation with the resulting increase in oxygen free radicals and destabilization of atherosclerotic plaques, may also increase the risk for ventricular electrical instability. In addition to the direct effects on myocardial structure, angiotensin II facilitates adrenergic effects on ventricular electrophysiology by reducing norepinephrine reuptake, increasing the amount of norepinephrine released with each nerve firing, and increasing the firing rate of sympathetic neurons [117–119] (Chap. 36).
Control system interactions operant in the heart failure milieu make it clear that the precise mechanisms involved in the development of lethal arrhythmias are complex and multifactorial. This illustrates that effective sudden prevention strategies must account for multiple potential mechanisms requiring the combination of multiple medications and device therapies.
7.3.5 Sympathetic Activation and Sudden Death (Chap. 5)
Sympathetic adrenergic activation in heart failure represents a combination of inherent control system characteristics present before heart disease develops coupled with how individual systems respond to ventricular injury [42]. Sympathetic activation is almost always accompanied by parasympathetic withdrawal, which should be considered when examining how autonomic alterations in heart failure lead to lethal arrhythmias.
In addition to functional changes in global sympathetic outflow to the heart, neural remodeling in the neuroeffector junction can be arrhythmogenic. Sympathetic hypersensitivity in areas of myocardial necrosis alters local cardiac electrophysiology increasing regional repolarization dispersion and increases risk for lethal arrhythmias [122, 123]. Myocardial injury can produce regional differences in nerve growth factors, which can stimulate sympathetic nerve “sprouting” and regionally increase relative sympathetic effect. This phenomenon further destabilizes ventricular electrophysiology and leads to increased risk for sudden death [120, 121].
Neural adverse remodeling relates to several factors, but the neural regeneration process after myocardial injury is mainly directed by growth factors, among which nerve growth factor (NGF) appears to be very important. Experimental data in conscious dogs documented that NGF infusion in the left stellate ganglion induces significant nerve sprouting (evaluated by analysis of the tyrosine hydroxylase (TH) and growth-associated protein 43 (GAP43 markers), leading to excessive expression of cardiac sympathetic innervation. This was directly associated with increased risk for sudden cardiac death [124–126].
Further elaboration of the impact of NGF in ischemic myocardium demonstrated that the factor is produced by cardiac myocytes and specific receptors for this neurotrophin (TrkA, tyrosine protein kinase receptor) are present in the myocyte membrane. NGF may have a beneficial role as it favors neoangiogenesis, which may provide some level of protection to ischemic myocardium [124]. These observations suggest that the process of cardiac nerve regeneration following myocardial injury may be an adaptive response designed to sustain failing cardiac contractility through sympathetic augmentation and angiogenesis, but at the maladaptive cost of higher risk of life-threatening arrhythmias.
The impact of adverse neural remodeling on cardiac electrophysiology is mediated through the development of anatomically fixed arrhythmogenic substrates that permanently disrupt orderly myocardial activation and repolarization characteristics of the heart. In addition, “functional” abnormalities, which are temporary and depend on local conditions such as acute sympathetic activation, myocardial ischemia, or inflammation, are directly mediated particularly through sympathetic remodeling. Functional abnormalities may include changes in local repolarization due to regional increases in sympathetic activation, for example, or may include activation fractionation from conduction delay induced by acute alteration in autonomic tone. Factors such as acute myocardial ischemia or infarction, electrolyte imbalances, or medications can also alter ion channel function resulting in high risk for functional electrophysiologic abnormalities that may lead to sudden death. Functional abnormalities tend to be difficult to predict and may occur shortly before the initiation of a sustained ventricular arrhythmia.
7.3.6 Cardiorenal Interactions and Risk for Lethal Arrhythmias
The presence of coexisting conditions has a substantial effect on the outcome of heart failure patients, and specifically kidney disease is associated with one of the highest risks of adverse outcomes driven both by heart failure progression and by lethal arrhythmia [126, 127]. This adverse interaction seems to relate to decreased renal perfusion as cardiac output drops in heart failure patients. Normally, the “renal fraction” is 20 % of cardiac output, but this percentage declines with worsening heart failure. This further enhances renal sympathetic efferent nerve activity that results in marked increases in renal norepinephrine spillover and a sympathetically mediated increase in plasma renin activity [128].
The kidney is robustly innervated by the sympathetic system and acts both as an afferent signaling source and as a conduit for increased sympathetic efferent activation [129], providing an important prognostic tool in evaluating and monitoring heart failure patients. Increased renal norepinephrine spillover predicts reduced survival in heart failure patients.
Activation of renal sensory nerves in patients with heart failure contributes to disease progression and triggers proarrhythmic cardiac substrate enhancing arrhythmia susceptibility [129]. This clearly impacts chronic kidney disease patient survival as lethal arrhythmias are a more frequent cause of death among this group even with moderate (stage 3) chronic kidney disease [131].
Worsening renal function in patients with heart failure is associated with more frequent lethal arrhythmias [129, 130]. A post hoc analysis of the MADIT II database suggests that an estimated glomerular filtration rate below 35 ml/min/1.73 m2 in ICD (implantable cardioverter defibrillator) patients was associated with a trend toward unfavorable outcome compared to the control group [131]. The reason ICDs were less effective in the low-GFR group seems related to the defibrillation threshold that progressively increases along with renal function decline. These factors lead to increased risk for arrhythmias [131]; at the same time defibrillation threshold makes ICD therapy less effective. Changes in myocardial tissue in the presence of renal dysfunction seem to be directly related to renal dysfunction which adversely impacts the arrhythmic substrate.
Other abnormalities, including electrolyte imbalances that may occur in the context of more intensive diuretic administration, are closely coupled with increased sympathetic activity. Adverse tissue remodeling in the presence of chronic kidney disease was evaluated in one study using cardiac magnetic resonance tomography in 24 hemodialysis patients. This data found abnormal patterns of late gadolinium myocardial tissue enhancement in 79 % of analyzed cases. Late enhancement was present in three distinctive patterns: a diffuse pattern was present in seven cases, an infarct-related pattern in six cases, a non-infarct related in seven cases, while the absence of structural remodeling was seen in only five cases [132]. The data suggest that a significant increase of myocardial interstitial fibrosis burden was induced by chronic renal failure. The study outlined some of the specific changes that take place in myocardial structure in patients with chronic kidney disease, which lead to proarrhythmic increase of defibrillation threshold.
In summary, neural and hormonal responses to heart failure result in permanent and functional alterations in myocardial activation and repolarization. The impact of neural and hormonal activation is rapid and continuous, which increases the risk for electrical “accidents” leading to sudden death. Understanding these mechanisms has led to very effective means to prevent sudden death, as well as the progression of myocardial dysfunction and heart failure.
7.4 Therapeutic Interventions: Drug Therapy
Further support for neurohormonal activation as a key mechanism leading to adverse tissue and electrical remodeling comes from clinical trials using specific drug therapies, such as beta-blockers [103, 106–108], ACE inhibitors, and aldosterone receptor antagonists [102, 133] (Table 7.1). Each of these interventions targets a specific component of the body’s response to heart failure and represents effective interventions to suppress lethal arrhythmias. ACE inhibitor therapy is known to reduce sudden death risk in patients with Stage A and B heart failure, which includes patients with coronary vascular disease or asymptomatic left ventricular dysfunction. However, angiotensin intervention with ACE inhibitors or angiotensin receptor blockers has more inconsistent effects on sudden death risk after heart failure develops. Beta-blocker therapy and aldosterone antagonism, however, consistently reduce the relative risk for sudden death in patients after myocardial infarction and in those with heart failure.
Pharmacologic interventions focus on functional or structural events that eventually lead to the development of lethal arrhythmias. Consequently, sudden death prevention in patients with chronic heart failure involves two basic mechanisms. Suppressing lethal ventricular tachyarrhythmias can be accomplished by preventing the fixed or functional electrophysiologic substrate conducive for arrhythmogenesis and suppressing electrophysiologic triggers responsible for initiating a reentrant circuit. Since the substrate for triggered activity and reentry is in large part dependent on neurohormonal activation in response to injury, it is not surprising that the most effective pharmacologic means to prevent lethal arrhythmias in patients with heart failure is with beta-blocker therapy coupled with angiotensin-converting enzyme inhibition and aldosterone antagonism (Chap. 53).
Such a concept was specifically tested in the CIBIS III trial in which bisoprolol titration occurred before ACE inhibitor therapy was started. This approach resulted in significantly lower sudden death mortality when compared to the standard guideline approach prescribing initial renin-angiotensin-aldosterone inhibition first before beta-blocker therapy [134].
Historically, the evolving understanding of functional and anatomic adverse remodeling of cardiac autonomic control has led to major therapeutic breakthroughs. After several years of study, it became apparent that beta-blockers were not contraindicated in heart failure patients, but actually a first-line therapy. Cardiac parasympathetic withdrawal occurs as heart failure progresses suggested by weakening of the baroreceptor reflex and low measurements of heart rate variability in patients with advanced heart disease. These observations led to the concept that direct vagal nerve or baroreceptor electrical stimulation may benefit patients with heart failure. Central integration of peripheral afferent signals in patients with heart failure may be altered by spinal cord electrical stimulation to attenuate the adverse changes in efferent neural output. These electrical stimulation therapies are the subjects of ongoing clinical trials in patients with heart failure [135, 136] (Chap. 6) (Table 7.2).
Table 7.2
Novel implantable electronic devices currently under development
Device | Action | Proposed cardiac effect in heart failure | SCD reduction |
|---|---|---|---|
Direct vagal nerve stimulation | Parasympathetic augmentation | Reverses adverse remodeling, prevents triggers for ventricular arrhythmias | Preclinical evidence for prevention. No clinical evidence |
Direct baroreceptor stimulation | Parasympathetic augmentation, sympathetic inhibition, central effect | Reverses adverse remodeling, prevents triggers for ventricular arrhythmias | Unknown |
Spinal cord stimulation | Alter central autonomic processing < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|