Right-to-Left Shunts
“Loeb’s Four Rules of Medicine
1. If the patient is getting better, keep doing what you are doing
2. If the patient is getting worse, change something
3. If you are not sure what you are doing, do nothing
4. Try to keep the patient out of the operating room”
Cyanosis is a physical sign characterized by a slate-blue color of the mucous membranes, nail beds, and skin. It results from the presence of deoxygenated hemoglobin in the blood at a concentration of at least 5 g per dL. At this or a slightly less concentration, deoxygenated hemoglobin may not be associated with cyanosis or may produce only a “ruddy” appearance. Cyanosis is less likely to occur in a patient with severe anemia because the hemoglobin levels may be too low to produce the color. In contrast, patients with erythrocythemia may exhibit a ruddy appearance or cyanosis despite a normal arterial partial pressure of oxygen (PaO2). Because cyanosis is a physical sign, detecting it depends on the observer’s acuity. It may be more difficult to appreciate the presence of cyanosis in patients who have heavily pigmented skin.
Hypoxemia is a state of abnormally decreased arterial blood oxygen concentration. It is recognized by measuring PaO2 or arterial blood oxygen saturation (SaO2). The degree of hypoxemia may or may not correlate with the physical signs of cyanosis, depending on the blood hemoglobin concentration and the ability of the examiner to detect cyanosis. A newborn, because of erythrocythemia, may appear plethoric or cyanotic despite a normal PaO2 and the absence of cardiac or pulmonary disease.
The relationship between PaO2 and SaO2 is important in understanding the determinants of cyanosis, hypoxemia, and tissue oxygenation. An arterial SaO2 of 88% (the level at which cyanosis is apparent to most observers) can be achieved with PaO2 ranging from 30 to 85 mm Hg, depending on the fetal hemoglobin concentration, pH, temperature, and level of 2,3-diphosphoglycerate (2,3-DPG).
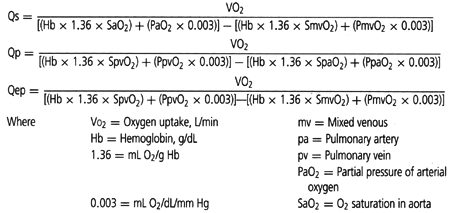
Normally, all the systemic venous return flows from the right atrium to the right ventricle (RV) and to the pulmonary artery. The volume of blood flowing into the pulmonary
artery is the same as the volume of blood that travels through the lungs to the left atrium and left ventricle and into the aorta. The volume of blood flowing into the aorta represents the systemic blood flow or Qs. The terms Qs and cardiac output are frequently used interchangeably. “Effective pulmonary blood flow,” or “Qep,” is the volume of systemic venous return that travels through the pulmonary vascular bed and becomes oxygenated (i.e., volume of pulmonary blood flow that is effective in participating in gas exchange in the lung). In the normal situation, the systemic, pulmonary, and effective pulmonary blood flows are equal—Qp = Qep = Qs. The formulae for calculating these volumes are listed in Table 10.1. The denominator of each of these equations merely describes oxygen extraction or addition to blood as it passes through either the systemic or the pulmonary vascular bed. For patients with cyanotic congenital heart disease, Qp, Qs, and Qep are usually not equal. The physiologic consequences of a specific defect can be understood and described by the values of Qp, Qs, and Qep and by the relationship among them.
artery is the same as the volume of blood that travels through the lungs to the left atrium and left ventricle and into the aorta. The volume of blood flowing into the aorta represents the systemic blood flow or Qs. The terms Qs and cardiac output are frequently used interchangeably. “Effective pulmonary blood flow,” or “Qep,” is the volume of systemic venous return that travels through the pulmonary vascular bed and becomes oxygenated (i.e., volume of pulmonary blood flow that is effective in participating in gas exchange in the lung). In the normal situation, the systemic, pulmonary, and effective pulmonary blood flows are equal—Qp = Qep = Qs. The formulae for calculating these volumes are listed in Table 10.1. The denominator of each of these equations merely describes oxygen extraction or addition to blood as it passes through either the systemic or the pulmonary vascular bed. For patients with cyanotic congenital heart disease, Qp, Qs, and Qep are usually not equal. The physiologic consequences of a specific defect can be understood and described by the values of Qp, Qs, and Qep and by the relationship among them.
TABLE 10.1 Tissue Oxygenation and Blood Oxygen Content | |
|---|---|
|
In addition, the concepts of left-to-right and right-to-left shunts are useful in understanding and describing the physiologic consequences of cyanotic forms of congenital heart defects. A left-to-right shunt exists when oxygenated blood from the lungs returns to systemic veins, the right atrium, the RV, or the pulmonary artery rather than going to the left atrium, left ventricle, and aorta. A right-to-left shunt exists when a volume of deoxygenated blood (systemic venous return) travels directly to the left atrium, left ventricle, or aorta instead of traveling through the lungs to be oxygenated.
An important concept in understanding the physiologic consequences of cyanotic forms of congenital heart defects is the relationship between the volume of pulmonary blood flow
and the degree of hypoxemia. Tetralogy of Fallot (TOF), pulmonary atresia with ventricular septal defect (VSD), truncus arteriosus, and tricuspid atresia are four defects in which the volume of pulmonary blood flow may be abnormally low and there is a communication between the two ventricles. In these instances, the lower the Qp, the more cyanotic the patient will be, and the higher the Qp, the less cyanotic the patient will be (see Fig. 10-1). Why is this? Consider TOF. SaO2 in the ascending aorta will depend on the relative volume of deoxygenated blood reaching the aorta from the RV and the volume of oxygenated blood reaching the aorta from the left ventricle. The volume of deoxygenated blood in the RV is relatively fixed and depends on systemic venous return to the atrium. However, the volume
of oxygenated blood in the left ventricle represents the volume of blood it receives from the left atrium, which in turn depends on the volume of pulmonary blood flow. In TOF, the volume of pulmonary blood flow depends on the degree of restriction of blood flow into the pulmonary artery from the right ventricular outflow tract obstruction.
and the degree of hypoxemia. Tetralogy of Fallot (TOF), pulmonary atresia with ventricular septal defect (VSD), truncus arteriosus, and tricuspid atresia are four defects in which the volume of pulmonary blood flow may be abnormally low and there is a communication between the two ventricles. In these instances, the lower the Qp, the more cyanotic the patient will be, and the higher the Qp, the less cyanotic the patient will be (see Fig. 10-1). Why is this? Consider TOF. SaO2 in the ascending aorta will depend on the relative volume of deoxygenated blood reaching the aorta from the RV and the volume of oxygenated blood reaching the aorta from the left ventricle. The volume of deoxygenated blood in the RV is relatively fixed and depends on systemic venous return to the atrium. However, the volume
of oxygenated blood in the left ventricle represents the volume of blood it receives from the left atrium, which in turn depends on the volume of pulmonary blood flow. In TOF, the volume of pulmonary blood flow depends on the degree of restriction of blood flow into the pulmonary artery from the right ventricular outflow tract obstruction.
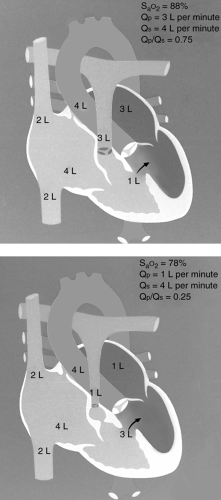 Figure 10.1 • Illustration of the relationship of the volume of pulmonary blood flow and systemic arterial blood oxygen saturation for a patient with tetralogy of Fallot. Top, the pulmonary blood flow is 3 L per minute and the systemic arterial blood oxygen saturation is 88%. Bottom, the pulmonary blood flow is only 1 L per minute and the systemic blood oxygen saturation is 78%. |
A second important concept is the anatomic relationship of the systemic veins (inferior and superior vena cavae and coronary sinus) and the pulmonary veins with the aorta and the pulmonary artery. Normally, the inferior and superior vena cavae are connected to the pulmonary artery through the right atrium and ventricle, and the pulmonary veins are connected to the aorta through the left atrium and ventricle. Deoxygenated blood is directed to the lungs, and oxygenated blood is directed to the body.
In d-transposition of the great arteries (d-TGA) (see Fig. 10-2), these relationships are unusual in that the inferior and superior vena cavae connect with the aorta through the right atrium and RV, and the pulmonary veins connect with the pulmonary artery through the left atrium and left ventricle. Therefore, deoxygenated blood is directed to the aorta, and oxygenated blood is directed to the pulmonary artery. Obviously, a communication must exist between these two circuits for survival. In this instance, the size of the communication and the degree of blood mixing through the communication determines the degree of cyanosis and hypoxemia, more than the volume of pulmonary blood flow. Indeed, in patients with TGA without a VSD, pulmonary blood flow, characteristically, is more than normal, but cyanosis is intense.
Total anomalous pulmonary venous return is another example of cyanotic congenital heart disease that results from an inappropriate relationship of the pulmonary venous drainage and the pulmonary artery (see Fig. 10-3). In this case, both the systemic and pulmonary venous return enters the pulmonary artery through the right atrium and RV.
With these concepts in mind, two questions must be answered to understand the anatomy and physiology of cyanotic forms of congenital heart disease: (i) What are the sources, reliability, and volume of pulmonary blood flow? and (ii) What is the relationship between the venous drainage to the heart and the arterial exit from the heart? The answers to these questions are critical in the clinical management of these patients.
Cardiac, pulmonary, metabolic, and hematologic diseases can produce cyanosis in the newborn. Any condition that causes cardiac shock can produce cyanosis, even in a patient with a structurally normal heart. When evaluating a cyanotic newborn, first consider the source of the cyanosis. Is it cardiac, pulmonary, metabolic, or hematologic (although methemoglobinemia rarely occurs in newborns) in origin?
The most common causes of cyanosis in newborns are cardiac and pulmonary diseases. Beginning with a thorough history, these two diseases must be distinguished. For a premature infant, respiratory distress syndrome is an important cause of cyanosis. If meconium aspiration occurred before or during delivery, pneumonia should be considered. In general, infants with cyanosis and pulmonary disease are more tachypneic and appear to have greater respiratory distress than those with cyanotic congenital heart disease, particularly in the first 24 to 48 hours of life. Newborns with cyanotic congenital heart disease are tachypneic but to a lesser extent than those with pulmonary disease. Significant pulmonary edema associated with a congenital cardiac defect can be misinterpreted as primary pulmonary disease. Many pulmonary causes of cyanosis are readily apparent from the chest radiograph, which should be obtained early in the evaluation of the cyanotic infant (see Table 10.2).
The use of echocardiography greatly simplifies the distinction of pulmonary from cardiac disease in infants. When echocardiography is not available, the “hyperoxia test” is useful and is performed by measuring and comparing PaO2 while the baby breaths room air and while the baby breaths 100% O2. It is important to measure the PaO2 and not just blood
oxygen saturation. For an infant breathing room air, PaO2 may be depressed to a similar level by pulmonary disease and by congenital heart disease. In cyanotic congenital heart disease, the PaO2 is decreased because of right-to-left shunting of blood; that is, systemic venous return (blood that is low in oxygen content) travels to the left side of the heart and into the aorta without going through the lungs to participate in gas transfer. Regardless of the
effectiveness of ventilation and the amount of oxygen delivered to each alveolus, the blood that is shunting from right to left cannot participate in gas transfer and cyanosis will persist.
oxygen saturation. For an infant breathing room air, PaO2 may be depressed to a similar level by pulmonary disease and by congenital heart disease. In cyanotic congenital heart disease, the PaO2 is decreased because of right-to-left shunting of blood; that is, systemic venous return (blood that is low in oxygen content) travels to the left side of the heart and into the aorta without going through the lungs to participate in gas transfer. Regardless of the
effectiveness of ventilation and the amount of oxygen delivered to each alveolus, the blood that is shunting from right to left cannot participate in gas transfer and cyanosis will persist.
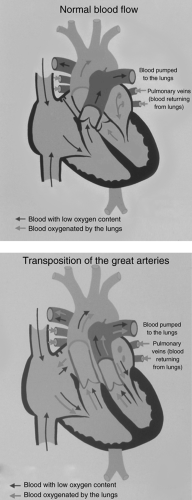 Figure 10.2 • Diagrammatic representation of d-transposition of the great arteries. |
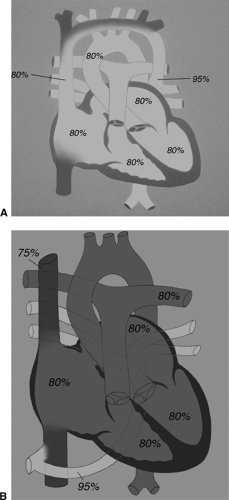 Figure 10.3 • Diagrammatic representation of (A) supracardiac totally anomalous pulmonary venous return (TAPVR) and (B) infradiaphragmatic TAPVR. |
In pulmonary disease, all the systemic venous return passes through the lungs, but, because of the pulmonary disease, not all the alveoli are ventilated appropriately. Blood passing through the unventilated alveoli cannot participate in gas exchange. However, if the fraction of inspired oxygen (FIO2) is increased and the patient is ventilated optimally, ventilation of these alveoli improves, gas exchange with the pulmonary blood flow improves, and PaO2 increases. As a rule of thumb, if PaO2 increases above 150 mm Hg when FIO2 = 1 and the baby is adequately ventilated, cyanotic cardiac disease is unlikely. However, if PaO2 fails to increase above 100 mm Hg, cyanotic congenital heart disease is probably the cause
of the cyanosis. If PaO2 is between 100 and 150 mm Hg, cardiac disease is likely to be the cause, but the diagnosis is uncertain.
of the cyanosis. If PaO2 is between 100 and 150 mm Hg, cardiac disease is likely to be the cause, but the diagnosis is uncertain.
TABLE 10.2 Pulmonary Causes of Cyanosis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Approach to Diagnosis
The goal of the primary care physician is to recognize the presence of significant heart disease in the infant. Utilizing the history, physical findings, chest radiography, and electrocardiography, one can make a reasonable assessment of the physiologic manifestations of the underlying congenital heart defect (increased or decreased pulmonary blood flow or presence or absence of a shunt). However, it is difficult to determine the underlying anatomic defect without the aid of echocardiography, cardiac catheterization, angiography, or a combination of these.
History
The family history and pregnancy and delivery history may be helpful in some cases. If there is a strong family history of congenital heart disease, one may be more likely to suspect this in the infant. Maternal diabetes or parental congenital heart disease is associated with a higher than normal incidence of the disease in the children. Perinatal problems such as meconium staining or aspiration may make pulmonary disease a more likely explanation than cardiac disease for cyanosis in the newborn.
Physical Examination
The respiratory pattern and frequency are helpful in distinguishing pulmonary from cardiac disease in newborns with cyanosis. The baby with cyanotic congenital heart disease will usually be tachypneic but will not have significant respiratory distress or retraction. An exception to this is the baby with obstructed totally anomalous pulmonary venous return (TAPVR) who will have significant respiratory distress resulting from pulmonary edema.
Careful palpation of the carotid, brachial, and femoral pulses is important in excluding associated coarctation of the aorta or interruption of the aortic arch. The quality of the precordial activity should be noted. Most forms of cyanotic congenital heart disease are associated with pressure or volume overload of the RV, and a prominent impulse will be apparent at the lower left sternal border or subxiphoid region.
The first heart sound is usually normal. The second heart sound is usually single in newborns with cyanotic congenital heart disease but, if widely split, Ebstein anomaly should be considered. The absence of a cardiac murmur does not exclude the presence of significant cyanotic congenital heart disease. A systolic pulmonary ejection murmur may suggest TOF; a harsh to-and-fro murmur would suggest absent pulmonary valve syndrome. A systolic murmur of tricuspid insufficiency and a tricuspid diastolic murmur may suggest Ebstein anomaly. A systolic murmur of tricuspid insufficiency can be heard in the infant if the delivery was stressful. In the newborn, the murmur of tricuspid insufficiency is easy to confuse with that of a VSD. However, it is unusual for the murmur of a VSD to be audible in the first 2 days of life when transient tricuspid insufficiency of the stressed newborn is heard.
Chest Radiograph
The chest radiograph is important in evaluating a newborn with cyanosis. It is most helpful in excluding noncardiac causes of cyanosis (Table 10.2). Except for certain classical appearances, the chest radiograph has limited use in defining the exact nature of a congenital cardiac defect. TGA often is associated with cardiomegaly and increased pulmonary vascular markings, but this is quite variable. The so-called egg on a string appearance of the cardiac silhouette is not very useful in the diagnosis of TGA (see Fig. 10-4). A chest radiograph showing a normal heart size and paucity of pulmonary vascular markings suggests TOF or pulmonary artery atresia with VSD, especially if there is also a right-sided aortic arch. Marked cardiomegaly and decreased pulmonary vascular markings suggests Ebstein anomaly or pulmonary atresia with intact ventricular septum.
Electrocardiogram
The electrocardiogram of a healthy newborn is characterized by right ventricular dominance. Left axis deviation and an initial counterclockwise frontal plane loop are abnormal and, in a cyanotic infant, are consistent with tricuspid atresia or complete atrioventricular septal defect with associated pulmonary stenosis (see Fig. 10-5).
d-Transposition of the Great Arteries
d-TGA is the most common form of congenital heart disease that presents with cyanosis in the newborn. It constitutes 3.8% of all congenital cardiac defects. d-TGA results from abnormal conotruncal septation such that the aorta arises from the RV and the pulmonary artery arises from the left ventricle. Forty percent of patients with d-TGA have an associated VSD. Among patients with d-TGA, 6% of those with intact ventricular septum and 31% of those with VSD have associated pulmonary stenosis.
In d-TGA, systemic venous return (blood with low oxygen content) is returned to the RV and is then pumped to the body through the aorta without passing through the lungs for gas exchange. The pulmonary venous return (oxygenated blood) to the left ventricle is pumped back to the lungs. The effective pulmonary blood flow (the volume of deoxygenated blood that participates in gas exchange in the lungs) is very low, although total pulmonary blood flow is increased. d-TGA is incompatible with life unless a communication exists between the two circuits to allow the mixing of oxygenated and deoxygenated blood. This mixture occurs at the patent foramen ovale (or atrial septal defect [ASD]), the ductus arteriosus (if patent), and the VSD (if present). This is a tenuous situation for a patient with an intact
ventricular septum and no true ASD because mixing of the two circuits will decrease as the patent ductus arteriosus closes and the patent foramen ovale becomes sealed.
ventricular septum and no true ASD because mixing of the two circuits will decrease as the patent ductus arteriosus closes and the patent foramen ovale becomes sealed.
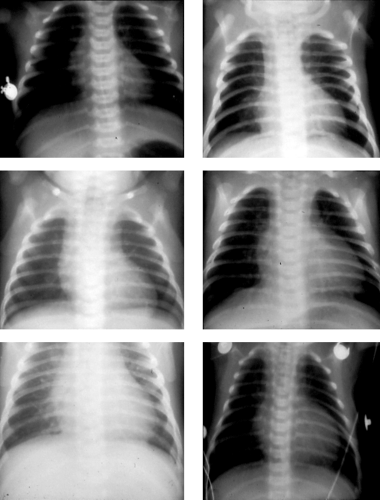 Figure 10.4 • Chest radiograms from six different infants with d-transposition of the great arteries. Note the great variability of the radiographs. The bottom left x-ray would be considered most typical. |
Cyanosis, a prominent precordial impulse at the lower left sternal border (“right ventricular impulse”), and a single and loud second heart sound are common findings in babies with d-TGA. There are no physical findings that are pathognomonic of d-TGA. There may
be no cardiac murmur or there may be a systolic ejection murmur, which can result from an associated VSD, pulmonary stenosis, or dynamic left ventricular outflow tract obstruction. Physical examination is important for excluding the presence of associated anomalies such as coarctation of the aorta.
be no cardiac murmur or there may be a systolic ejection murmur, which can result from an associated VSD, pulmonary stenosis, or dynamic left ventricular outflow tract obstruction. Physical examination is important for excluding the presence of associated anomalies such as coarctation of the aorta.
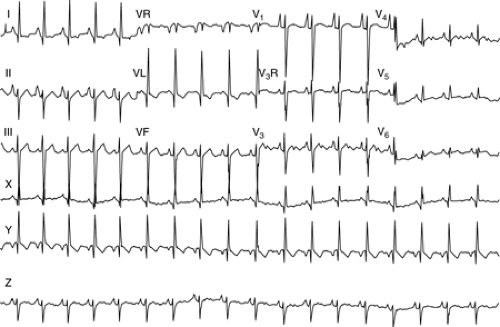 Figure 10.5 • An electrocardiogram demonstrating left axis deviation and atrial enlargement. This would be most characteristic of tricuspid atresia. |
The chest radiograph shows cardiomegaly with increased pulmonary vascular markings in most patients with d-TGA and intact ventricular septum. Patients with d-TGA, VSD, and pulmonary stenosis may have decreased pulmonary vascular markings.
A newborn with d-TGA represents a medical emergency. It is critical to establish or exclude this diagnosis and to document and ensure adequate sites for mixing between the parallel systemic and pulmonary circuits of d-TGA. The diagnosis of d-TGA and most associated malformations can be established using two-dimensional echocardiography. The presence of a patent ductus arteriosus can be determined, but the ductus arteriosus cannot be relied upon as a stable source of mixing because it likely will close. The ductus arteriosus can be maintained patent by infusion of prostaglandin E1, and this should be done if the baby is extremely hypoxemic (PaO2 <25 mm Hg), acidotic, or to be transferred to another institution.
Management of d-Transposition of the Great Arteries with no Ventricular Septal Defect
Arterial Switch (Jatene) Procedure
This procedure represents the best option for managing infants with d-TGA, intact ventricular septum or VSD, and a normal pulmonary valve. In this operation, the aorta and pulmonary artery are transected cephalad to their respective valves. The ostia of the coronary arteries
are removed from the stump of the aorta and sewed to the stump of the pulmonary artery (the neoaorta). The distal portion of the aorta is anastomosed to the proximal stump of the pulmonary artery, and the distal portion of the pulmonary artery is anastomosed to the stump of the aorta (see Fig. 10-6). The mortality for this procedure is 10% or less, and the long-term results are quite good. This procedure must be done early (<3 weeks of age),
before the thickened left ventricular walls involute as pulmonary arterial resistance decreases. In many institutions, the operation is performed without preoperative cardiac catheterization. Prostaglandin E1 is infused preoperatively to maintain patency of the ductus arteriosus and preserve the acid-base stability of the patient.
are removed from the stump of the aorta and sewed to the stump of the pulmonary artery (the neoaorta). The distal portion of the aorta is anastomosed to the proximal stump of the pulmonary artery, and the distal portion of the pulmonary artery is anastomosed to the stump of the aorta (see Fig. 10-6). The mortality for this procedure is 10% or less, and the long-term results are quite good. This procedure must be done early (<3 weeks of age),
before the thickened left ventricular walls involute as pulmonary arterial resistance decreases. In many institutions, the operation is performed without preoperative cardiac catheterization. Prostaglandin E1 is infused preoperatively to maintain patency of the ductus arteriosus and preserve the acid-base stability of the patient.
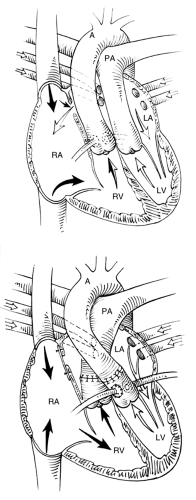 Figure 10.6 • Diagrammatic representation of the arterial switch (Jatene) operation for d-transposition of the great arteries. Note the suture lines that connects the proximal stump of the pulmonary artery to the aorta and the second suture connecting the stump of the aorta to the pulmonary artery. A, aorta; PA, pulmonary artery; LA, left atrium; LV, left ventricle; RV, right ventricle; RA, right atrium. |
Atrial Switch (Senning or Mustard) Procedure
The Senning and Mustard operations were described in the 1960s and predate the arterial switch operation. These are alternatives to the arterial switch procedure. They are used if the pulmonary valve is abnormal such that it would not work as the neoaortic valve following a Jatene (arterial switch) procedure. With these procedures, systemic and pulmonary venous returns are rerouted in the atrium. The systemic venous return from the superior and inferior vena cavae is directed through the mitral valve and into the left ventricle (and subsequently to the pulmonary artery). The pulmonary venous return is directed through the tricuspid valve (and subsequently to the aorta). This is accomplished by sewing a baffle in the atrium (see Fig. 10-7). In essence, TGA is treated by creating “transposition” of the venous return to the heart.
The Senning and Mustard operations usually are performed in patients 6 months to 1 year of age, but some surgeons will perform the operation in the newborn period. If one of these methods of operation is to be utilized, an adequate interatrial communication must be established in the newborn period. This is accomplished by a Rashkind balloon atrial septostomy. A catheter with an inflatable balloon at its tip is advanced from the femoral or umbilical vein to the right atrium through the patent foramen ovale and into the left atrium. The balloon is inflated in the left atrium, and withdrawn rapidly into the right atrium, producing a rent in the atrial septum.
The Senning and Mustard operations have a low operative mortality but significant intermediate and long-term problems. These include obstruction of systemic and pulmonary venous return by the baffle, atrial arrhythmias, tricuspid insufficiency, and right ventricular failure.
Management of d-Transposition of the Great Arteries with Ventricular Septal Defect
When a significant VSD coexists with d-TGA, the systemic and pulmonary venous returns can mix through the VSD. In general, babies with associated VSD but without pulmonary stenosis are less hypoxemic than those with intact ventricular septum. d-TGA and VSD without pulmonary stenosis can be managed by an arterial switch procedure (Jatene) plus VSD closure. The presence of a large VSD prevents left ventricular pressure and wall thickness from declining as pulmonary resistance decreases. Therefore, in contrast to patients with d-TGA and no VSD, the arterial switch procedure can be delayed until 1 to 2 months of age.
Management of d-Transposition of the Great Arteries with Ventricular Septal Defect and Pulmonary Stenosis or Atresia
The physiology of d-TGA with VSD and pulmonary stenosis is different from that of d-TGA without pulmonary stenosis because pulmonary blood flow is limited by the pulmonary stenosis. The degree of hypoxemia in these patients will be determined both by the extent of mixing of the systemic and pulmonary venous returns and by the volume of pulmonary blood flow. In these patients, the sources and adequacy of the pulmonary blood flow must
be determined. With severe pulmonary stenosis or pulmonary artery atresia, the ductus arteriosus may be contributing significantly to the pulmonary blood flow. When the ductus arteriosus closes, severe hypoxemia and acidosis may ensue. In the presence of an abnormal pulmonary valve, these patients are not candidates for an arterial switch (Jatene) procedure. Eventually, repair is best accomplished using the Rastelli procedure, which involves closing the VSD so that blood exits the left ventricle through the VSD and enters the aorta. The pulmonary artery is divided, the proximal stump is oversewn, and a conduit is placed from the RV to the distal portion of the main pulmonary artery. This operation has the disadvantage of using a prosthetic extracardiac conduit, which eventually will fail and need to be replaced. To allow the placement of a reasonably large conduit, this operation usually
is delayed until age 4 to 6 years. Before 4 to 6 years of age, it may be necessary to establish a systemic-to-pulmonary artery shunt to augment pulmonary blood flow.
be determined. With severe pulmonary stenosis or pulmonary artery atresia, the ductus arteriosus may be contributing significantly to the pulmonary blood flow. When the ductus arteriosus closes, severe hypoxemia and acidosis may ensue. In the presence of an abnormal pulmonary valve, these patients are not candidates for an arterial switch (Jatene) procedure. Eventually, repair is best accomplished using the Rastelli procedure, which involves closing the VSD so that blood exits the left ventricle through the VSD and enters the aorta. The pulmonary artery is divided, the proximal stump is oversewn, and a conduit is placed from the RV to the distal portion of the main pulmonary artery. This operation has the disadvantage of using a prosthetic extracardiac conduit, which eventually will fail and need to be replaced. To allow the placement of a reasonably large conduit, this operation usually
is delayed until age 4 to 6 years. Before 4 to 6 years of age, it may be necessary to establish a systemic-to-pulmonary artery shunt to augment pulmonary blood flow.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


