Fig. 13.1
Diagram representing the intermediate role of arterial stiffness in chronic cardiorenal syndromes. See text for explaining separate depicted, and additional amplifying components of the triangle
Arterial stiffness is defined as a reduction in arterial distensibility. Several indices of a stiffened arterial system have all been associated with dismal cardiovascular outcome [4]. In CKD, aortic stiffness is an important additional risk factor for mortality [5]. Moreover, a wide pulse pressure (the difference between systolic and diastolic blood pressure) itself may induce glomerular damage [6, 7]. Clinically, arterial stiffness can be measured using sophisticated software programs that analyse arterial pressure curve on a single site like the brachial or radial artery, using pulse wave velocity (PWV), measuring time interval between arrival of systolic peak pressure at two sites with different distance from the left ventrical, or by estimating arterial stiffness by the difference between the systolic and diastolic blood pressure (pulse pressure) [8]. A stiffened central artery like the aorta has crucially different physical properties compared to a physiologically compliant artery. This translates into a completely different hemodynamic profile in the cardiovascular system during the cardiac cycle. The energy or power generated by the left ventricle during systole can be regarded as the sum of the blood volume replacement, i.e. the stroke volume, and pressure increment, the systolic blood pressure. When the compliance of the compartment where this energy is replaced to (the aorta) declines, more of this energy will comprise of pressure workload instead of volume, due to lack of capacity to dilate [9, 10]. In physiological situations a part of energy transfer from the left ventricle is “stored” as elastic expansion of central arterial structures. In stiffened central arteries this is not possible. Initially this will lead to an increase is systolic pressure and a decline in peripheral blood supply. The latter in turn will lead to a higher demand from the left ventricle to meet global blood flow requirements, and induce hypertrophy. A second physical feature of a stiffened artery is an increased velocity of the pressure wave generated during systole at the opening of the aortic valve. It is important to differentiate this pressure wave from the actual propagation of blood volume through the aorta, the latter being much slower. Since the pressure wave is reflected from the smaller peripheral arterial system, it traverses back in the opposite direction from the blood flow. With stiffened arteries, this reflected pressure wave reaches the aortic root more early, and eventually, as arterial stiffening progresses, close to the peak pressure of the cardiac cycle, as such augmenting peak pressure in the central arteries (Fig. 13.2). This induces an increase in cardiac afterload, and is an additional trigger for the development of LVH.
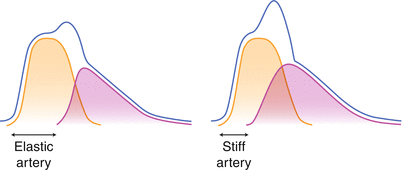
Fig. 13.2
Pressure curves measured in time form left to right in the proximal aorta during one cardiac cycle. Left panel: normal compliant central arterial system shows two components of the pressure curve. In Orange the hypothetical curve, assuming the absence of a reflected pressure curve. This curve is the sole consequence of antegrade pressure transduction form the left ventricle during systole. In Purple the pressure curve from the pressure reflection by more distal resistant arterial system that has traversed back retrogradely to the proximal aorta. The Blue line represents the actual pressure which is the sum of the two separate pressure curves. Right panel: Although the shape of the two pressure curve do not differ substantially the purple reflected curve returns more early in the cardiac cycle due to higher speed of pressure transduction along a stiffened arterial system. As a consequence the actual peak pressure (the bule line) is much higher: pressure augmentation. The additional height of the peak pressure, on top of the peak of the orange curve is referred to as the augmentation index
The elastic recoil of the distended proximal aorta after systole is an important driver of coronary perfusion during diastole, the time period in which myocardial perfusion is most optimal due to the relaxation of the myocardial wall [11, 12]. The loss of recoil capacity of stiffened arteries, in the presence of LVH, may induce a severe mismatch between metabolic needs of the hypertrophied myocardium and blood supply to that tissue, although to some extent this is attenuated by coronary autoregulation.
Arterial Stiffness
As outlined, stiffening of central arteries can be an important intermediate phenomenon linking cardiovascular risk factors in CKD to the increased prevalence of related clinical complications [13]. Therefore, at least hypothetically, arterial stiffness could be an attractive target for specific inventions to prevent these complications. This raises the fundamental question of reversibility of this phenomenon in CKD. It is important to underline that in CKD vascular pathology can differ from non-CKD subject by the presence of extensive calcification of the tunica media, a complication most likely to have detrimental consequences for arterial distensibility. Aiming to reverse arterial stiffness can conceptually be subdivided into attempts to either improve the quantity of vessel calcification, and to target functional and structural vascular changes that CKD patients have in common with non-CKD subjects with arterial stiffness. Clinically there are no methods to weigh the relative contribution of these two components, but examining the different pathophysiological processes, as will be outlined in the subsequent section, may form the basis of complementary therapeutic approaches, if available.
Vascular Calcification
It is clear that the patient in the introduction has a stiffened arterial system, based on the high pulse pressure and the increased PWV of 15 m/s. Normal values of PWV have been firmly established and should be indexed to age and actual blood pressure [14]. Facing the fact this patient has CKD and possibly insulin resistance raises the possibility that the high PWV may be partly attributed to vascular calcification [15]. In CKD a typical form of calcification exists, called Monckeberg’s sclerosis, which is located in the medial layer, the site responsible for the elastic and contractile properties of the arterial wall. Clinically, this type of calcification can be suggested using several radiological techniques, including sophisticated computed tomography or plain X-ray techniques such as lateral abdominal views (Fig. 13.3). These techniques do not differentiate between calcified atheromatous plaques of the intima, or medial calcification. However, it can be expected that any calcification, regardless of its histological location, interferes with arterial distensibility, but the impact on stiffness is probably higher for the medial calcification, because it is more continuous in localisation and not patchy like calcified plaques. If present, the questions arise if progression of this calcification can be halted or even reversed, and ultimately if this modification of calcification benefits the patient.
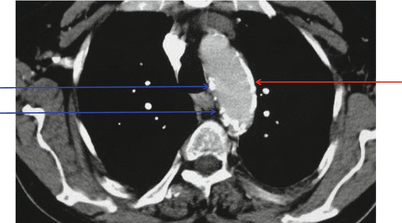
Fig. 13.3
CT scan of the thorax at the level of the arcus aortae, showing a large plate-like calcified area (red arrow) and some areas with more patchy distribution of calcified regions (blue arrows) (Courtesy of professor David Goldsmith)
When considering rational intervening in the process of vascular calcification, in-depth knowledge of its pathogenesis is required. The central player appears to be the vascular smooth muscle cell (VSMC) that undergoes a phenotypic switch when exposed to a uremic milieu, with key roles for abnormal levels of calcium and phosphate [16]. A complex interplay between deranged minerals, abnormal VSMC’s, deranged gene expression profiles, unbalanced local calcification inhibitors and promotors, and deregulated endocrine networks all are involved in the development of calcification. Several of these components are modifiable, but clinically meaningful regression of established vascular calcification has not been shown [17]. Nonetheless, there are clues that some strategies might at least slow the progression of calcification. Active vitamin D appears protective against VSMC-mediated calcification [18] but caution is warranted since local actions of active vitamin D from plaque-infiltrating macrophages might worsen arterial stiffness [19], and its beneficial role is debated [20, 21]. Use of phosphate-binder therapy is generally applied in more advanced CKD, when phosphate concentrations exceed normal values. The rationale for its use is the central role of phosphate in the pathogenesis of calcification, and the fact that phosphate itself is a structural component of hydroxyapatite, the structure that calcified laesions are made of. Overall, phosphate binder therapy does not appear to influence vascular calcification [22, 23] or at best slow its progression in dialysis patients with the possible exception of calcium-containing binder that may even accelerate it [24]. Besides phosphate, a central role for calcium in cardiovascular calcification is evident. Indeed, calcium channel blocker therapy, besides being an antihypertensive drug, appears to attenuate coronary artery calcification [25]. Therefore, additional manipulations targeting calcium could theoretically interfere with the calcifying propensity in CKD. In dialysis patients, the use of the calcimimetic cinacalcet tended to slow the progression of calcification in the thoracic aorta, but this was not significant, and the compound cannot be used in more early stage CKD [26]. More importantly, it is clear that to slow progression is not to regress it and only regression could improve this component in arterial stiffness.
Recent insights point to the role of matrix gla protein (MGP), and its relation with vitamin K, which is required for activating the mineralisation-inhibiting properties of MGP. Especially in the setting of vitamin K deficiency or inhibition of its activity by the use of coumarin-therapy, this could be an important tool for intervention by either stopping coumarin or supplementing vitamin K [27, 28]. Although Fetuin A may be as important as MGP as calcification inhibitor, its role is not completely elucidated as levels do not parallel calcification burden in CKD, possibly because of initial upregulation as a defence mechanism against calcification [29]. The role of novel treatments, originally developed for the treatment of bone disease like bisphosphonates and RANK-L inhibitors (denosumab) is currently far from elucidated with possible some beneficial effects in older subjects using bisphosphonates, but no effect of denosumab [30, 31]. In conclusion, currently available pharmacological intervention are not proven effective in reversing calcification, beneficially influence vascular stiffness or lead to improved clinical outcome. However, novel insights into molecular mechanism of medial calcification do hold promise for future targeted interventions (Fig. 13.4).
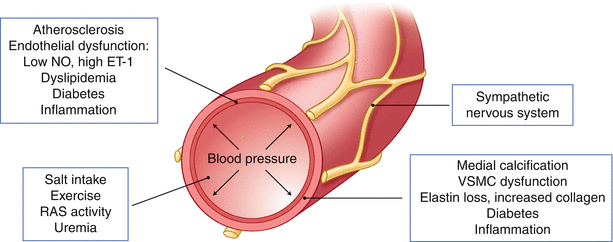
Fig. 13.4
Diagram representing well-established factors that contribute to arterial stiffness, grouped into four categories: primarily affecting the medial layer (right lower panel), intimal layer (left upper panel), distant or humoral and endocrine factors (left lower panel) and the sympathetic nervous system (right upper panel)
Arterial Stiffness Not Related to Vascular Calcification
Despite having an increased risk for vascular calcification the patient in the introduction has several features that are associated with arterial stiffness, not attributable to calcification. Higher age, hypertension, diabetes (besides being a risk factor for vascular calcification), and high salt intake could all contribute to arterial stiffness, and could potentially be modified, except for age. To what extent CKD per se contributes to this causes of arterial stiffness is unknown, since many factors that are related to low arterial distensibility in the non-CKD population frequently aggregate in CKD patients. Data for instance of change in PWV in acute renal failure or following nephrectomy are lacking. However, since these non-calcifying modulators of arterial stiffness are highly prevalent in CKD, they are an integral component of treatment for these patients.
Since arterial stiffness frequently occurs in the absence of vascular calcification, it is clear other causes are operating too. Indeed, physical, structural and functional components can contribute to this vascular rigidity, and each could be modifiable [32]. From Fig. 13.2 it is clear that a direct relation between PWV and blood pressure exist. Since there is a limit to the maximum elastic expansion of central arterial structures, it is obvious that at higher central blood pressure these vessels are distended more, and as such are more close to the maximum distensibility and more rigid.
In understanding the relation between blood pressure and arterial stiffness it is crucial to realise that the blood pressure measured at the brachial artery may differ substantially form that in the aorta, and that the effects of antihypertensive agents may have different blood pressure effects centrally versus more peripheral like the brachial artery [33]. Especially inhibitors of the renin-angiotensin system (RAS) have a more pronounced central blood pressure lowering effect [34], possibly explaining the finding that despite similar peripheral blood pressure lowering effect, these compounds reduce PWV more than other blood pressure lowering drugs [35]. At the same peripheral blood pressure, those with a reduced central blood pressure had fewer cardiovascular events and declining renal function, as shown in a subset of the ASCOT study, that were treated by combination therapy using amlodipine and perindopril [36]. As inhibitors of the RAS, calcium channel blockers have consistently been shown to improve indices of arterial stiffness [37]. The reasons for the diverging effect on central blood pressure of several classes of antihypertensive drugs could either be a direct effect on the vessel wall itself or a replacement of the virtual reflection point of the reversed pressure pulse, and as such lowering the central pressure augmentation by the reflected wave. The latter may be of less importance since LVH correlates better with PWV than with augmentation index, once blood pressure is controlled [38]. In addition to the physical effects of blood pressure on arterial stiffness, chronic exposure to hypertension changes the structure of the vessel wall. Especially the loss and integrity of elastin and increased content of collagen may contribute to increased stiffness. Long-term treatment of hypertension may improve these structural changes to some extent [39]. Especially for the mineralocorticoid receptor antagonists like spironolacton and eplerenone it is suggested that their beneficial effects on arterial stiffness are better explained by an improvement in arterial structure than by their blood pressure lowering effects [40]. In addition to hypertension several other factors have been implicated to induce these non-calcification structural changes in the arterial wall. Among these are salt-intake (beyond its effect on blood pressure) [41], and possibly low intake of fish-oil [42], low levels of physical exercise and obesity.
The recognition that arterial tone is not only dependent on blood pressure and structural components in the matrix, like elastin, collagen and the presence of atheromatous plaques and medial calcification, pointed to functional properties that to a large extent depend on the interplay between endothelial cells and VSMC. Although the latter may play a more important role more distally in the arterial tree, compared to the more elastic most central artery, the aorta, the role of vascular cellular function on arterial stiffness is likely important [43]. Vascular tone is partly dictated by several endothelial derived substance like the vasodilator nitric oxide (NO) and the vasoconstrictor endothelin-1. Endothelin-A receptor inhibitors may improve arterial stiffness in CKD-patients when accompanied by a decline in ADMA (asymmetric dimethylarginine) and improve their risk profile, and hold promise for the future [44]. Statins, beside its well-known effects as cholesterol-lowering drugs, also improve arterial stiffness, an effect that too may be mediated by its effect on endothelial cells [45]. This potential beneficial effect of statins has also been clearly shown in CKD-patients, and even in patients on dialysis a minimal decline in PWV (of very doubtful clinical benefit) has been shown [46, 47]. In part due to the technical progress made in renal nerve ablation the role of the sympathetic nervous system (SNS) on arterial stiffness in physiology, which is an increase in PWV with increased sympathetic activity, has gained interest. [48, 49] Recently it was shown that renal denervation for hypertension in both non-CKD and CKD patients indeed showed marked improvement in arterial stiffness (measured by decline in augmentation index), which was independent from blood pressure lowering effects [50, 51]. As overactivity of the SNS in CKD is almost universal, targeting its activity may have unexpected beneficial effects on vascular stiffness, beyond possible hypertension improvements. Technically less challenging, though sometimes more difficult to achieve is the cessation of smoking. Arguably none of all described factors that deteriorate arterial stiffness have been so consistently described as smoking. Importantly, parameters of arterial stiffness all improve with time after smoking cessation [52].
As for its role in vascular calcification, the beneficial effects of vitamin D on vascular stiffness is debated. In a large observational study it was shown that in the general population low levels of cholecalciferol (the nutritional non-active form) were associated with more arterial stiffness [53]. Results of a placebo-controlled intervention study supplementing cholecalciferol was less convincing: though central blood pressure decreased no change was noticed in PWV [53], while in older subject with isolated systolic hypertension or postmenopausal women no effect at all was noticed [54, 55].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


