13 OBJECTIVES
Respiratory Failure
![]() Delineate and understand the symptoms and management of respiratory failure.
Delineate and understand the symptoms and management of respiratory failure.
![]() Evaluate clinical presentations and coordinate appropriate mechanical ventilation therapies.
Evaluate clinical presentations and coordinate appropriate mechanical ventilation therapies.
![]() Be familiar with the risks associated with instituting mechanical ventilation and complications that may result.
Be familiar with the risks associated with instituting mechanical ventilation and complications that may result.
![]() Analyze and comprehend a clinical pathway for evaluating and treating a patient with respiratory failure with the help of clinical scenarios.
Analyze and comprehend a clinical pathway for evaluating and treating a patient with respiratory failure with the help of clinical scenarios.
GENERAL CONSIDERATIONS
 Respiratory failure, whether acute or chronic, remains one of the most common life-threatening medical problems encountered in critical care. It is estimated that 34% of patients in critical care in the United States receive mechanical ventilation. Short-term survival rates for patients with acute respiratory failure (ARF) that is not preceded by preexisting disease or associated multiorgan failure are more than 85%. Hospital mortality rates for acute exacerbations of chronic obstructive pulmonary disease (COPD) are less than 20%; however, quality of life and 1-year survival rates are generally poor. Outcomes of patients with ARF with preexisting disease depend primarily on the underlying conditions. Approximately 17% of patients placed on mechanical ventilation require support for more than 14 days.
Respiratory failure, whether acute or chronic, remains one of the most common life-threatening medical problems encountered in critical care. It is estimated that 34% of patients in critical care in the United States receive mechanical ventilation. Short-term survival rates for patients with acute respiratory failure (ARF) that is not preceded by preexisting disease or associated multiorgan failure are more than 85%. Hospital mortality rates for acute exacerbations of chronic obstructive pulmonary disease (COPD) are less than 20%; however, quality of life and 1-year survival rates are generally poor. Outcomes of patients with ARF with preexisting disease depend primarily on the underlying conditions. Approximately 17% of patients placed on mechanical ventilation require support for more than 14 days.
Respiratory failure is a functional disorder caused by conditions that impair the ability of the respiratory system to remove carbon dioxide from and deliver oxygen to the pulmonary capillary bed. This inability to complete gas exchange results in abnormally low arterial oxygen tension with or without abnormally high arterial carbon dioxide tension.
The clinical manifestations of respiratory failure are linked to the patient’s ability to adapt to hypoxemia and hypercapnia. The concept of cardiopulmonary reserve is vital to understanding a patient’s ability to adapt. Cardiopulmonary reserve refers to the interdependence of the heart, lungs, and oxygen-carrying capacity of any given patient. A patient with an impaired cardiac pump, preexisting pulmonary disease, or abnormalities in hemoglobin will have less “reserve” and will require a more urgent intervention for a respiratory event in comparison to a patient with normal physiology. Normal lungs, heart, and hemoglobin permit a degree of physiologic reserve that allows patients to compensate for an acute respiratory insult.
ETIOLOGY & PATHOGENESIS
Respiratory failure may be acute or chronic, depending on when the process develops, with varying pathogenic, physiologic, and therapeutic distinctions. Both acute and chronic respiratory failure can be classified into two main groups distinguishable by arterial blood gas analysis. The first category, hypoxic respiratory failure (HRF), is characterized by an abnormally low partial pressure of arterial oxygen (Pao2) with a normal or low partial pressure of arterial carbon dioxide (Paco2). The second category, hypercapnic-hypoxic respiratory failure (HHRF), is associated with an abnormally elevated Paco2 coupled with an abnormally low Pao2.
Acute Hypoxic Respiratory Failure
Hypoxic respiratory failure may be defined as any pulmonary condition resulting in severe arterial hypoxemia (Pao2 <50 mm Hg) that cannot be corrected by increasing the fraction of inspired oxygen (Fio2) to >50% (>0.5). Pathophysiologic causes of hypoxemia with an anatomic correlation are outlined in Table 13–1A and B Acute respiratory failure limited to hypoxemia alone generally represents a disease process that involves only the lung, as it functions in gas exchange. Severity of ventilation-perfusion mismatching correlates well with the inability to increase Pao2 despite subsequent increases in supplemental oxygen. Typically, the alveolar-arterial gradient increases with the addition of inspired oxygen, and the ratio of Pao2:Fio2 (PF ratio) becomes increasingly low.
Table 13–1A. Causes of hypoxemia: Anatomic correlation with pathophysiological derangement
Anatomic | Pathophysiology |
Airspace filling defect |
|
Interstitial inflammation and fibrosis | Diffusion impairment |
Vascular lesions/shunts | Shunt defects |
Airway compromise or collapse | Hypoventilation |
 mismatch
mismatchClinical Diagnosis | ABGs |
COPD, chronic asthma | Reduced Pao2, increased Pco2 |
Diffuse interstitial pulmonary disease | Reduced Pao2, variable Pco2 |
Congestive heart failure, pulmonary embolism | Reduced Pao2, reduced Pco2 |
UAO, OSA, morbid obesity, chest wall defects | Reduced Pao2, increased Pco2 |
Note: COPD, chronic obstructive pulmonary disease; UAO, upper airway obstruction; OSA, obstructive sleep apnea.
Acute respiratory distress syndrome (ARDS) is the clinical correlate of a severe form of acute lung injury (ALI) that causes acute and persistent lung inflammation with increased capillary permeability. ARDS can occur without other organ damage as in severe inhalational injury or air embolism, and the early pathologic features are described as diffuse alveolar damage accompanied by the release of proinflammatory cytokines and toxic mediators, causing endothelial damage and acute lung injury (ALI). Alveolar and interstitial edema occurs due to protein and fluid shifts that overwhelm the capacity of the lymphatics. As a result, the air spaces fill with bloody, proteinaceous edema fluid and debris from degenerating cells with loss of surfactant and subsequent atelectasis. This in turn causes impaired gas exchange due to ventilation-perfusion abnormalities and severe right-to-left arteriovenous shunts with decreased compliance due to poorly aerated and stifflungs. Further hypoxic vasoconstriction and vascular compression causes pulmonary hypertension. A chronic state may result in secondary erythrocytosis and increased blood viscosity leading to right ventricular strain and cor pulmonale (Figure 13–1). ARDS progresses through three relatively discrete pathologic stages. The initial “exudative” stage, characterized by diffuse alveolar damage, is followed by a “proliferative” stage, which is characterized by resolution of pulmonary edema and by proliferation of type II alveolar cells, squamous metaplasia, and interstitial infiltration by myofibroblasts, and early deposition of collagen. Some patients progress to a third “fibrotic” stage, characterized by obliteration of normal lung architecture, diffuse fibrosis, and cyst formation.
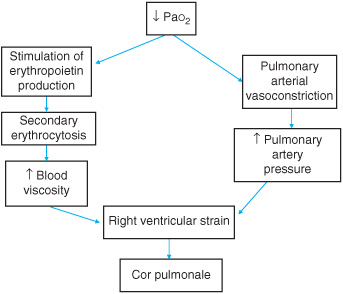
Figure 13–1. Consequences of chronic hypoxic respiratory failure.
Hypercapnic-Hypoxic Respiratory Failure
Hypercapnic-hypoxic respiratory failure is best defined as a ventilatory insufficiency resulting from a reduction in minute ventilation, or an increase in dead space ventilation that is associated with a direct reduction in alveolar ventilation.
A specific level of Paco2 attributable to HHRF is difficult to assign without knowledge of the cause of the respiratory failure and the patient’s previous condition. During hypoventilation, the carbon dioxide pressure (Paco2) and Pao2 levels change in opposite directions by nearly the same amount (Figure 13–2). HHRF is always associated with some degree of hypoxemia. Even with a severe degree of alveolar hypoventilation, resulting in a doubling of Paco2, hypoxemia will not be a dominant feature in the normal lung due to a normal alveolar-arterial oxygen gradient. In contrast there is worsening hypoxemia in a diseased lung insulted with diminished alveolar ventilation. The primary etiology of HHRF is impaired gas exchange that results in increased dead space, ventilation-perfusion mismatches, and reductions in alveolar ventilation. The partial pressure of carbon dioxide within the alveoli (Paco2) is increased and the Pao2 is subsequently reduced.
In emphysema, the pathogenesis is thought to be secondary to destruction of the alveolar capillary interface. The mechanism is primarily a function of tobacco use, which causes excess oxygen stress with alveolar macrophage and neutrophilic inflammation damaging the alveolar septum. This results in loss of the vascular bed and lung recoil with marked overinflation, causing derangement of gas exchange and dead space ventilation. Bronchial asthma is the second most common cause of HHRF. There is increased large and small airway resistance secondary to mast cell and eosinophilic inflammation with edema, airway remodeling, mucus hypersecretion, and muscle hypertrophy. HHRF may result from the failure of the structures that contribute to the ventilation of the lung, including central nervous system disorders, neuromuscular disorders, and fatigue. (See Chapter 6 for further discussion.) The primary mechanism for hypoxemia in HHRF secondary to COPD and asthma is the perfusion of poorly ventilated lung units or ventilation-perfusion mismatch. The clinical manifestations of respiratory failure are dictated by difficulties in gas transport and alveolar ventilation. The symptom complexes are linked to the patient’s adaptability to changes in hypoxemia and hypercapnia. The degree of ventilatory response is more closely correlated with the capacity of the respiratory system to respond to hypoxemia than with its ability to respond to hypercapnia. As a direct result of hypoxemia, the sympathetic nervous system is activated, creating a direct outflow of catecholamines. The cardiovascular response to catecholamine stimulation is tachycardia, increased systemic vasoconstriction, and increased cardiac output. Arterial hypoxemia directly stimulates the carotid body chemoreceptors, leading to an increase in minute ventilation. In cases of severe hypoxemia the myocardium and central nervous system are at greatest risk of injury; tissue hypoxia is associated with impaired mental performance and myocardial dysfunction. Clinical manifestations of HHRF are compounded by the presence of hemato-logic or circulatory abnormalities. Acute hypercapnia causes central nervous system depression by lowering cerebrospinal fluid pH. The rapid decreases in serum pH, as opposed to absolute levels of serum Paco2, best correlate with a depression in mental status. Hypercapnia does stimulate increases in minute ventilation in normal subjects, but this response is commonly blunted by the pathology that led to the episode of HHRF. This diminished response impairs effective improvement in alveolar ventilation.
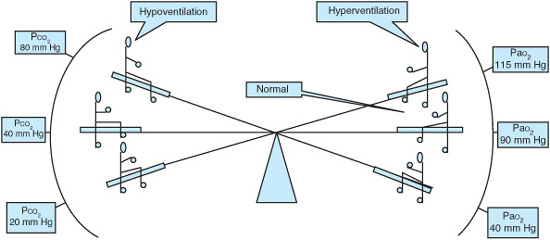
Figure 13–2. Relationship between PaO2 and PaCO2: Illustrative examples.
DIAGNOSTIC & CLINICAL CONSIDERATIONS
Evaluation should initially focus on the events leading to the onset of respiratory distress, such as drug effect, infection, trauma, or neurologic events such as seizures, as well as any underlying disease such as asthma and COPD. Findings are dependent on the cause of respiratory failure and ARDS. These may include fever, hypotension, abdominal pain, or coma.
Common clinical causes of HRF are listed in Table 13–2 and include but are not limited to pneumonia, aspiration, sepsis, trauma, neurologic insults, and post-transplant lung injuries. Massive blood transfusions and leukoagglutinin reactions can cause severe acute lung injury, ARDS, and lead to respiratory failure.
In ARDS, respiratory distress usually follows 24–48 hours after some systemic insult, with rapidly worsening tachypnea, dyspnea, and hypoxemia requiring high concentrations of supplemental oxygen; dry cough and chest pain may also be present. The physical examination usually reveals cyanosis, tachycardia, tachypnea, and diffuse rales in the chest. Laboratory findings are nonspecific and may include increased white cell count, decreased platelets, evidence of disseminated intravascular coagulation (DIC), and metabolic acidosis or lactic acidosis. Arterial blood gases usually show an acute respiratory alkalosis, an elevated alveolar-arterial oxygen gradient and severe hypoxemia. A metabolic acidosis is often present in patients with sepsis and shock. The hypoxemic patient will appear agitated, and at times delirious (Table 13–3). As a direct result of hypoxemia, the sympathetic nervous system is activated, creating a direct outflow of catecholamines. The cardiovascular response to catecholamine stimulation is tachycardia, increased systemic vasoconstriction, and increased cardiac output. Arterial hypoxemia directly stimulates the carotid body chemoreceptors, leading to an increase in minute ventilation. In cases of severe hypoxemia the myocardium and central nervous system are at greatest risk of injury; tissue hypoxia is associated with impaired mental performance and myocardial dysfunction. It is important to identify the underlying cause (Table 13–4).
Table 13–2. Causes of acute hypoxic respiratory failure (HRF)
Adult respiratory distress syndrome (ARDS) |
Severe acute respiratory syndrome (SARS) |
Aspiration |
Atelectasis (lobar) |
Cardiogenic pulmonary edema |
Lung contusion or alveolar hemorrhage |
Pneumonia |
Pneumothorax |
Pulmonary embolus |
Transfusion-related acute lung injury |
Table 13–3. Symptoms of hypoxemia and hypercapnia
Hypoxemia | Hypercapnia |
Tachycardia | Somnolence |
Tachypnea | Lethargy |
Anxiety | Restlessness |
Diaphoresis | Tremor |
Altered mental status | Slurred speech |
Confusion | Headache |
Cyanosis | Asterixis |
Hypertension | Papilledema |
Hypotension | Coma |
Bradycardia |
|
Seizures |
|
Lactic acidosis |
|
Although HHRF is always associated with some degree of hypoxemia, specific causes of HHRF are listed in Table 13–5. HHRF may result from the failure of the structures that contribute to the ventilation of the lung, including central nervous system disorders, neuromuscular disorders, and fatigue.
In patients with HHRF the chest radiograph often shows significant hyper-inflation in asthma and COPD, or small lung volumes in neuromuscular disease and drug overdose.
Table 13–4. Causes of Adult respiratory distress syndrome (ARDS)
Aspiration |
Fresh and salt water |
Gastric contents |
Hydrocarbons |
Central nervous system |
Anoxia |
Increased intracranial pressure |
Seizures |
Trauma |
Drug overdose or reactions |
Acetylsalicylic acid |
Cocaine |
Heroin and other opiates |
Paraquat |
Plaquenil |
Propoxyphene |
Hematologic alterations |
Disseminated intravascular coagulation |
Massive blood transfusion |
Leukoagglutination reactions |
Infections |
Pneumonia (bacterial, fungal, viral) |
Sepsis |
Tuberculosis |
Inhalation injury |
Corrosive chemicals (NO2, Cl2, NH3, phosgene) |
Oxygen |
Smoke |
Metabolic disorders |
Pancreatitis |
Uremia |



