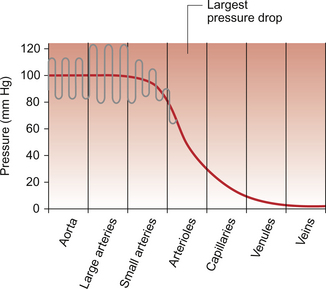
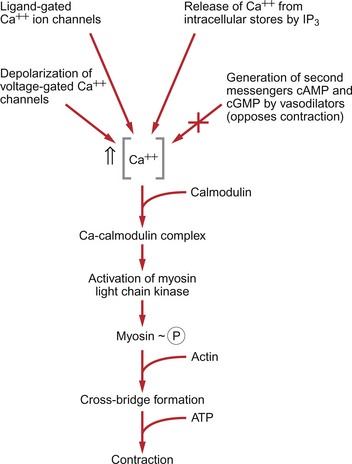
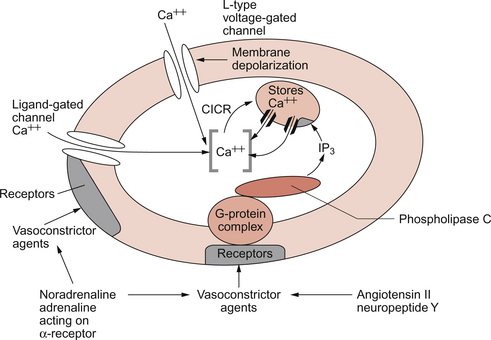
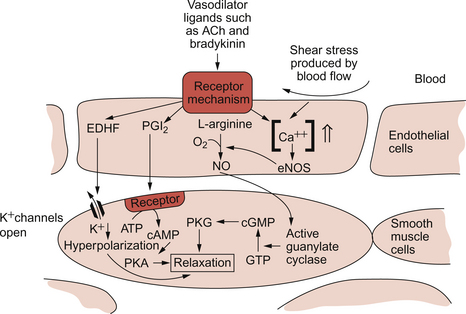
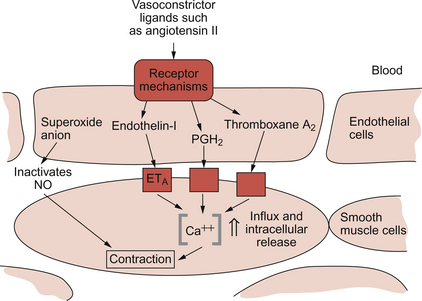
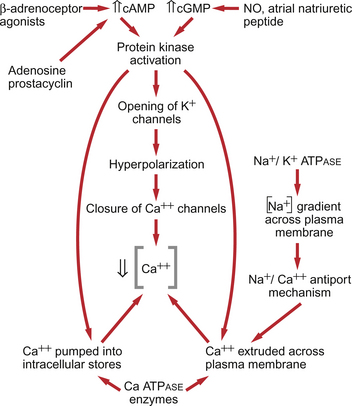
9 Failure to match blood flow to metabolic need will lead to loss of function of a tissue, to the development of pain and subsequently to tissue death. The case history of a lady with a problem in the regulation of the blood flow to her fingers is introduced in Case 9.1:1. Where the blood flows to when it leaves the aorta depends on the relative resistance to flow in each part of the circulation. In each case, the blood flows through a series of blood vessels, arteries, arterioles, capillaries, venules and veins. The structure of the walls of all these vessels is described in Chapter 1 (see Figs 1.7, 1.8). As noted in Chapter 8, there has to be a pressure gradient to achieve blood flow. Mean pressure in the arterial tree is typically close to 100 mm Hg and pressure in the right atrium is about 0 mm Hg (i.e. close to atmospheric pressure). Figure 9.1 shows the pressure drop going round the systemic circulation and it can be seen that the population of blood vessels through which there is the largest drop in pressure is the arterioles. These vessels, therefore, must be the segment of the circulation that have the highest resistance to blood flow. This concept is important because it means that by regulating the arterioles, we can: The product of the cardiac output and the peripheral resistance to blood flow determines the arterial blood pressure (see Chapter 10). As the arterial pressure provides the driving force to perfuse tissues, physiological control systems act to keep arterial pressure relatively constant from moment to moment and from day to day. Indeed, sustained raised arterial pressure can cause serious damage to many parts of the body (see Chapter 10). The consequence of a fall in arterial pressure is often poor brain blood flow which results in syncope (fainting). Adjustment of the arteriolar resistance is achieved by altering the state of contraction of vascular smooth muscle. The mechanisms of smooth muscle contraction are now described. Smooth muscle is located in the walls of the hollow structures of the body including blood vessels, airways, gut and bladder. The cells are spindle-shaped with a central nucleus and this is the first way in which smooth muscle cells differ from skeletal or cardiac muscle cells (see Chapter 6). As with the other two types of muscle, contraction of smooth muscle is triggered by a rise in intracellular [Ca++]. The source of the calcium is, however, different in the three types of muscle. The calcium involved in skeletal muscle contraction is stored intracellularly. It is released from the sarcoplasmic reticulum and is pumped back into these stores during muscle relaxation. In cardiac muscle, most of the calcium used in contraction derives from intracellular stores but some enters the cardiac muscle cell down a concentration gradient from the extracellular fluid via plasma membrane calcium ion channels (see page 20). The membrane potential of vascular smooth muscle studied in vitro is close to −60 mV but in vivo it is only about −40 mV. This is because the pressure inside blood vessels stretches the smooth muscle and this stretch leads to the opening of a population of ion channels which result in partial depolarization of the cell and hence partial contraction of the smooth muscle. This is the basis for what has long been known as the ‘Bayliss Effect’. Basically, if you stretch vascular smooth muscle it responds by contracting. An advantage of having partially contracted vascular smooth muscle is that physiological mediators (locally released chemicals, hormones or neurotransmitters) can either cause further contraction or relaxation of smooth muscle as appropriate. Some physiological mediators (see section ‘Metabolite control of local blood flow’ on p. 104) act via a population of ATP-sensitive K+ channels. A decrease in [ATP] inside the smooth muscle cell increases the probability that this population of K+ channels will be open. This leads to hyperpolarization and hence relaxation of smooth muscle. Relaxation of smooth muscle requires that intracellular [Ca++] is reduced. This can be achieved either by pumping the calcium back into intracellular stores or by expelling it outside the cell (see page 20 for a description of the equivalent mechanisms in cardiac muscle). The use of different sources of calcium for contraction in the three types of muscle is illustrated by the pharmacological effects of calcium channel blocking drugs. Drugs such as nifedipine, diltiazem and verapamil will, to varying degrees, reduce heart rate and the contractility of the heart (see Chapter 5). These drugs may also be used to achieve peripheral vasodilatation as part of antihypertensive therapy (see Chapter 10). Their side effects are fairly predictable. These include facial flushing, headache and dizziness as a result of their effects on vascular smooth muscle but also constipation is a common side effect because of the effects of calcium channel blocking drugs on gut smooth muscle. Calcium channel blocking drugs have no effect on skeletal muscle function because all the calcium needed for contraction is stored within the sarcoplasmic reticulum. The calcium channel blocking drug nifedipine was tried as therapy for the patient with Raynaud’s disease described in Case 9.1:2, the aim being to cause vasodilation and improve the blood flow to the fingers. The contractile mechanism for smooth muscle is different to the two other types of muscle. In skeletal and cardiac muscle the contractile proteins, actin and myosin, are arranged in parallel layers and this is the origin of the striated (striped) appearance when these muscles are viewed under the polarized light microscope. Contraction of striated muscle (see Chapter 2) is initiated by the binding of Ca++ to the control protein troponin. This has the effect of moving another protein, tropomyosin, out of a groove on the bundle of actin filaments. Formation of a ‘cross-bridge’ is then achieved by the myosin head having access to a binding site on the actin filament. Muscle contraction takes place with the hydrolysis of ATP to provide the energy. Smooth muscle does have actin and myosin as contractile proteins but does not have troponin. The Ca++ released into the cytosol of smooth muscle cells binds to the protein calmodulin. The calcium–calmodulin complex activates the enzyme myosin light chain kinase and this promotes phosphorylation of the myosin filament. Once this has been achieved, interaction between actin and myosin phosphate generates contraction of the smooth muscle cell. Figure 9.2 summarizes the events associated with cross-bridge formation and hence contraction of smooth muscle. When intracellular [Ca++] decreases, myosin is dephosphorylated by myosin light chain phosphatase. Even when dephosphorylated myosin can retain its interaction with actin. These attachments are called latch-bridges. They only detach slowly and so they maintain a level of muscle tension with little consumption of ATP. There are several broad types of mechanism which contribute to the overall regulation of intracellular [Ca++]. These mechanisms are illustrated in Figures 9.3 and 9.4. Some vasoconstrictor agents such as noradrenaline (norepinephrine) act through more than one mechanism: Fig. 9.4 Main pathways leading to a decrease in intracellular [Ca++] as part of vasodilatation mechanisms. • Vasoconstrictor hormones such as noradrenaline, angiotensin II, endothelins, vasopressin and thromboxane A2 bind to G-protein coupled receptors. Subsequent generation of the second messenger inositol trisphosphate (IP3) leads to the opening of channels in intracellular calcium stores and release of Ca++ (Fig. 9.3). • Vasoconstrictors also lead to membrane depolarization by several mechanisms. These include opening of ligand gated ion channels in the plasma membrane which permits influx of Na+ and Ca++ accompanied by inhibition of K+ channels (Fig. 9.3). • Intracellular [Ca++] also depends on the Ca++ removal mechanisms. These include pumping Ca++ back into intracellular stores and active extrusion of Ca++ across the plasma membrane both of which involve Ca-ATPase enzymes. There is also a Na+/Ca++ antiport exchanger. Entry of Na+ into the cell down its concentration gradient is coupled to extrusion of Ca++ against its concentration gradient. The low intracellular [Na+] is of course maintained by the sodium pump (Na+/K+ ATPase) (Fig. 9.4). • Vasodilator agents act via production of either cAMP (e.g. adenosine, prostacyclin, β-adrenoceptor agonists) or cGMP (nitric oxide, atrial natriuretic peptide) as second messengers. Both cAMP and cGMP activate protein kinases and hence lead to protein phosphorylation. A reduction in plasma [Ca++] may then be secondary to cell hyperpolarization following opening of K+ channels. The hyperpolarization closes Ca++ channels. An alternative mechanism for vasodilatation is the activation of Ca++ pumps leading to either extrusion of Ca++ from the cell or sequestration of Ca++ into intracellular stores (Fig. 9.4). Inappropriate spasm of vascular smooth muscle is the diagnosis suggested in the case study in Case 9.1:2. The adult human circulation consists of about 60 000 miles of tubing (see Chapter 1). It is lined by a thin monolayer of endothelial cells. These cells not only provide a barrier between the blood and the other cells of the body (see Chapter 11), but they are also the source of a range of vasoactive agents which cause relaxation or contraction of underlying blood vessel smooth muscle (Figs 9.5, 9.6). One of these compounds, before it was chemically identified, was initially named endothelium-derived relaxing factor (EDRF). It is now thought that most, but not necessarily all, of the vascular effects of EDRF can be attributed to nitric oxide. Other factors produced by the endothelium and which also affect vascular smooth muscle contraction have been identified (see p. 103). NO is synthesized from the amino acid l-arginine by the action of nitric oxide synthase (NOS) enzymes (Fig. 9.5). The terminology for these enzymes is a little confusing as it reflects the original site of discovery rather than current opinion of their site of importance. Endothelial NOS (eNOS) and neuronal NOS (nNOS) are both constitutively expressed in a wide range of cells, including many cell types in the cardiovascular system. These enzymes generate NO continuously. Inducible NOS (iNOS) is synthesized by cells exposed to inflammatory cytokines such as tumour necrosis factor alpha (TNFα), interleukin 1β (IL1β) and interferon alpha (IFNα). A range of other cytokines have the opposite effect and suppress iNOS expression. Overall the balance of local cytokines determines the expression of iNOS and the rate of NO production as required. NO produced in this way in macrophages has cytotoxic actions. Excessive production of NO by iNOS also occurs in some forms of septic shock and will lead to peripheral vasodilatation and a fall in arterial blood pressure (see Chapter 14). It is assumed that iNOS generated NO does not contribute to the normal physiological control of blood vessel diameter.
RESISTANCE BLOOD VESSELS
Introduction
Resistance to blood flow
Vascular smooth muscle
Source of Ca++ for smooth muscle contraction
Contraction of smooth muscle
Local control of vascular smooth muscle
Endothelial factors in the control of local blood flow
Nitric oxide (NO)
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree