Vessel type
Artery
Mean α1-AR density (fmol/mg protein)
Arteries
Central
Aorta
9.8 ± 1.8
Small somatic
Mammary
6.0 ± 1.3
Splanchnic
Mesenteric
13 ± 6.1
Splenic
28 ± 7.7
Hepatic
28 ± 5.9
Renal
Renal
23 ± 5.9
Pulmonary
Pulmonary artery
24 ± 5.2
Coronary
Epicardial coronary
2.1 ± 0.67
Veins
Central
Vena cava
16 ± 2.8
Small somatic
Saphenous
10 ± 1.2
Renal
Renal vein
18 ± 5.8
Pulmonary
Pulmonary vein
18 ± 4.6
The neural innervation of the lung ultrastructure is complex and involves a network of efferent, sensory, and neurohumoral pathways. Amine Precursor Uptake and Decarboxylation (APUD) cell clusters, which express catecholamine synthesis potential, are also distributed throughout the pulmonary vascular bed [5]. Whereas APUD-regulated catecholamine control of epithelial smooth muscle cell function is linked to the development of airway disease, such as asthma, the influence of these cells to the pathogenesis of pulmonary vascular disease is less well defined. Nevertheless, pulmonary vascular/lung nerve stimulation is associated with frequency-dependent increases in pulmonary artery perfusion pressure [6], while surgical or chemical denervation of proximal pulmonary arterioles influences pulmonary artery and RV systolic pressure directly in experiment pulmonary hypertension in vivo [7]. In one single center study of 13 PAH patients, changes from baseline at 3 months following pulmonary artery denervation were noted for mean pulmonary artery pressure (mPAP) (55 ± 5 vs. 36 ± 5 mmHg, p < 0.01), 6-min walk distance (6-MWD) (324 ± 21 vs. 491 ± 38 m, p < 0.01), and Tei index (e.g., assessment of RV systolic function) (0.7 ± 0.04 vs. 0.5 ± 0.04, p < 0.01) [8], providing preliminary data to support future investigations assessing the validity of this strategy for use in clinical practice.
Despite these collective observations, studies quantifying neurohumoral activation in PAH patients report variable conclusions. Plasma venous norepinephrine levels correlated inversely in PAH (N = 60) with cardiac output (r = −0.29, p < 0.05) in one study [9], which is a finding supported by other data demonstrating an association between circulating norepinephrine levels (N = 21) and pulmonary artery pressure (r = 0.66, P < 0.01), cardiac index (r = −0.56, P < 0.01), and pulmonary vascular resistance (r = 0.69, P < 0.001) [10]. Sympathetic over-activation in PAH is supported further by work from Velez-Rosa and colleagues [11], who analyzed muscle sympathetic nerve activity (MSNA) in 17 PAH patients. They observed that MSNA, which measures electrical activity of the peroneal nerve fascicle using tungsten microelectrodes to assess muscle-directed sympathetic nerve trafficking, was increased by 33 % (P < 0.01) in PAH patients compared with controls. A significant, inverse relationship between MSNA and exercise capacity, assessed by New York Heart Association functional class, was also observed.
Adrenergic Signaling and PAH
The G-protein coupled AR subtypes β1 and β2, are expressed constitutively in human pulmonary artery endothelial cells (PAECs) in a 1:3 distribution [6]. Stimulation of β1 and β2 receptors, in turn, is linked to endothelium-dependent regulation of pulmonary vascular tone. For example, extra-lobar pulmonary arteries dissected from mice and treated with isoproterenol (10−9–10−5 mol/L), the selective β2-AR agonist procaterol (0.1 μM), but not the β3 agonist CL316243, demonstrated a significant pulmonary vascular relaxation response. In turn, this effect was attenuated by inhibition of endothelial nitric oxide synthase (eNOS) with L-NG-Nitroarginine Methyl Ester (L-NAME) (300 μM) or selective pharmacological antagonism of the β1– or β2– AR, and was abolished fully in transgenic mice lacking eNOS [12].
Perturbation to the redox balance of RV cardiomyocytes and pulmonary vascular cells is also implicated in abnormal β-AR-nitric oxide (NO•) signal transduction. In cultured pulmonary artery endothelial cells, peroxynitrite (ONOO−) oxidizes eNOS to decrease bioavailable levels of NO•, which disrupts pulmonary vascular tone and promotes adverse remodeling. Increases in RV cardiomyocyte ONOO− levels assessed with anti-nitrotyrosine immunohistochemistry are observed in pulmonary hypertensive rats and are associated with RV dysfunction and decreased cardiac output in vivo [13]. In turn, pertussis toxin restores β-AR signaling in pulmonary artery smooth muscle cells [14], suggesting that Gi uncoupling induced by ONOO- may be an important mechanism by which β-AR-NO• signaling is disrupted under conditions of increased pulmonary vascular oxidant stress, as has been reported in various forms of pulmonary vascular disease.
In contrast to the beneficial effects of β-AR signal transduction on RV and pulmonary vascular function, α-AR stimulation is linked to adverse pulmonary arterial remodeling and the development of pulmonary hypertension. For example, stimulation of α1-AR in PASMCs and adventitial fibroblasts by norepinephrine promotes a maladaptive pulmonary vascular hypertrophy induced by hypoxia in pulmonary hypertensive mice [15]. In turn, transgenic mice deficient in epinephrine and norepinephrine synthesis or α1-AR expression appear protected from luminal occlusion and intimal thickening under the same conditions [15]. The determinates of pathogenic α1-AR stimulation are largely unknown, but may involve cross-talk between α- and β-AR signaling, particularly with respect to α1-AR-dependent regulation of β-AR chloride (Cl−) conductance, which has been shown to inhibit β-AR signal transduction targets [16].
Adrenergic Signaling in PAH: The Right Ventricle
Pharmacological modification of catecholamine signal transduction through β-, α-, and D-receptor antagonism or antagonism is a bona fide treatment strategy for patients with various stages of heart failure due to left ventricular dysfunction. Despite the routine use of these therapies for this purpose, the differential effect of manipulating catecholamine function on RV performance in patients with left-sided or right-sided heart failure is not well established. Furthermore, the extent to which changes in adrenergic receptor expression or activity promotes RV dysfunction under conditions of increased RV afterload, such as in pulmonary hypertension, is incompletely characterized.
Piao and colleagues recently addressed these areas of uncertainty by characterizing changes to the β-AR- and D1-signaling profiles in various models of experimental RV dysfunction in vivo [17]. They observed a greater decrease in RV cardiomyocyte α1– and β1-AR protein/mRNA expression levels in the hypertrophied RV under conditions of chronic inflammatory pulmonary vascular injury compared to acute pulmonary occlusion through pulmonary artery banding. These data are in concert with earlier studies demonstrating that compared to control hearts or patients with biventricular heart failure, β1-AR expression levels are decreased in the RV of PAH patients with isolated right heart failure [18, 19]. The authors assert that this finding distinguishes, in part, adaptive from maladaptive RV hypertrophic remodeling, respectively, as pulmonary hypertension was common to both experimental models. In that study, intracellular cAMP production was attenuated and activity of G protein–coupled receptor kinase-2 (GRK2), which is implicated in adrenergic receptor down regulation in LV cardiomyocytes, was increased in the maladaptive RV hypertrophied hearts. Collectively, these findings were in support of additional findings indicating RV cardiomyocyte β1-AR desensitization in PAH and the potential superiority of inotropic agents that maintain adenylyl cyclase coupling under these conditions, including dobutamine to preserve contractility in the failing RV.
These and similar observations from others [20] suggest that strategies to modulate RV hypertrophy directly may offset abnormal adrenergic signaling patterns in PAH. Several investigators have explored this to observe that pharmacological inhibition of α1-/β1-/β2-signaling improves RV structure and performance in experimental PAH without differentially affecting pulmonary vascular function in vivo. Bogaard and colleagues [21] reported that although no significant change was noted in pulmonary arterioles of Sugen-5416/hypoxia-PAH rats treated with carvedilol, drug treatment promoted RV reverse remodeling and improved tricuspid plane annular excursion (TAPSE), and increased cardiac output. This effect was associated with decreased RV thickness and intramyocardial fibrosis burden, as well as alterations to the molecular signature of the hypertrophied RV vis-á-vis cardiomyocyte fetal gene activation, including decreased α-myosin heavy chain gene and increased fetal β-myosin heavy chain expression levels. Similar findings are observed for the selective β1-AR antagonist bisoprolol, which decreased RV thickness and improved ventricular elastance in one study of monocrotaline-PAH [22], while the beneficial effects of selected other AR antagonists on RV function is also reported under alternative experimental conditions such as high altitude and hypobaric hypoxia [23].
Translating AR Axis Interventions to PAH Clinically
Reconciling the favorable effects of β-AR stimulation in RV or pulmonary vascular tissue (e.g., increased NO• bioactivity) under normal conditions with benefits linked to β-AR antagonism in experimental PAH is an important area of active investigation. It is interesting to note that AR signal transduction regulates interleukin 6 (IL-6) and vascular endothelial growth factor (VEGF) [24], which are both involved in pulmonary endothelial injury and recapitulation of PAH experimentally. It is also possible that dysregulated catecholamine-AR signaling in PAH is a consequence of pathogenic changes to intermediaries downstream of AR activation. For example, the β1-AR target sarcoplasmic reticulum Ca2+-ATPase 2a (SERCA2a), which regulates intracellular Ca2+ content in vascular smooth muscle cells, is down regulated in PASMCs and pulmonary arterioles in monocrotaline-PAH [25], while gene transfer via intratracheal delivery of aerosolized adeno-associated virus serotype 1 (AAV1) carrying SERCA2a reverses pulmonary endothelial dysfunction, decreases PAP, vascular remodeling, and RV hypertrophy significantly in PAH in vivo [25].
It is possible that the functional consequences of β-AR signaling on pulmonary vascular function vary in the intact versus injured pulmonary endothelium. Overall, the balance of available data points to dysregulated adrenergic signaling in pulmonary vascular and RV tissue in PAH, although the identification of factors responsible for mediating a transition from healthy to maladaptive AR signaling in PAH remain incompletely characterized. Along these lines, consensus is pending regarding the risk-reward balance of AR-modulating pharmacotherapeutics clinically. While attempts to replicate in PAH/RV dysfunction patients the (substantial) benefits of AR antagonists observed in left heart disease, this strategy has been evaluated empirically only in small observational studies limited largely to patients with selected forms of pulmonary vascular disease (e.g., portopulmonary hypertension) and in whom treatment was with early generation drugs (propranolol or atenolol) at high doses [26]. Some PAH experts maintain reservations regarding the safety profile of AR antagonists [27], particularly owing to the association of this drug class with diminished functional capacity due to chronotropic/RV inotropic insufficiency in patients with (exercise-induced) pulmonary vascular disease, while recently published expert consensus guidelines referenced insufficient clinical data to outline formal recommendations for their use in practice [28]. To bridge this knowledge gap, several randomized, prospective clinical trials have been announced assessing the effect of AR antagonists, including carvedilol, on outcome in PAH (NCT00964678; NCT01723371; NCT01586156; NCT0225394 at clinicaltrials.gov) (Table 9.2), while further efforts are also required to clarify optimal AR agonist therapy in the critically ill PAH patient with severely impaired cardiac output.
Table 9.2
Registered clinical trials investigating the effect of therapies that influence neurohumoral and/or hormonal signaling on outcome in pulmonary arterial hypertension (PAH)
Study name | Identifier/duration | Treatment | Inclusion criteria | Endpoint | Primary secondary |
|---|---|---|---|---|---|
Beta-blockers for the treatment of PAH in children | NCT01723371 6 mo End date: 9/2014 | Carvedilol | Age: ≥8 and ≤17.5 yr mPAP >25 mmHg PCWP <15 mmHg PVR >3 Wood units Clinically stable (3 mo) | Adverse event incidence | Δ 6MWD, VO2 Δ TAPSE Δ RVEF |
Beta-blockers in PAH | NCT01246037 6 mo End date: 4/2014 | Bisoprolol | Age: ≥18 yr Stable iPAH WHO Class II/III | ΔRVEF by CMR Safety measures | RV diastolic function Sympathetic activity levels |
PAH treatment with carvedilol for heart failure (PAHTCH) | NCT01586156 6 months End date: 7/2018 | Carvedilol | Age: 18–65 yr PAH WHO Class I-III | ΔHIF/NO/ AR recovery ΔRV function | |
Pilot study of the safety and efficacy of carvedilol in PAH | NCT00964678 6 mo End date: 5/2014 | Carvedilol | Age: ≥18 yr WHO Group 1 PH NHYA Class II/III mPAP >25 mmHg 6MWD >100 m | ΔRVEF by CMR | ΔRVESV Δ6MWD ΔTAPSE |
Spironolactone for PAH | NCT01712620 6 mo End date: 11/2015 | Spironolactone | Age: ≥18 yr WHO Group 1 PH Stable therapy (4 wk) mPAP >25 mmHg PCWP <15 mmHg PVR >3.0 Wood units NYHA Class I-III | Δ6MWD Clinical worsening | ΔVO2 ΔRV function Inflammation biomarkers Drug safety |
Effects of spironolactone on collagen metabolism in patients with PAH | NCT01468571 16 wk End date: 12/2015 | Spironolactone | Age: ≥18 yr Body weight >40 kg WHO Group 1 PH Stable therapy (4 wk) | ΔFibrosis markers | Adverse events Δ6MWD ΔFunctional class Clinical worsening |
Modulating effects of lisinopril on sildenafil activity in PAH (MELISSA) | NCT01181284 32 wk End date: 7/2011 | Lisinopril added to sildenafil | Age: 18–75 yr WHO Group I PAH PVR >3 Wood units PCWP ≤16 6MWD 150–575 m PDE-Vi (3 mo) | ACE-I tolerability | ΔN-BNP levels ΔGas exchange measures Δ6MWD |
Combined ambrisentan plus spironolactone in patients with pulmonary arterial hypertension (CAPS-PAH) | NCT0225394 6 mo End date: 6/2017 | Spironolactone added to ambrisentan | Age: 16–75 yr WHO Group I PAH PVR >3 Wood units PCWP ≤16 6MWD 150–575 m Ambrisentan/PDE-Vi (3 mo) | Combined: (a) 6-MWD (b) pVO2 | Fibrosis markers RV function QOL |
Hormonal, metabolic, and signaling interactions in PAH | NCT01884051 5 yr End date: 9/2017 | ACE-2 Metformin | Sex hormone metabolite levels Δ6MWD Glucose metabolism | Hemodynamics PET scan results |
Aldosterone as a PAH Disease Modifier
Elevated levels of aldosterone in lung has been demonstrated in monocrotaline- and Sugen-5416/hypoxia-PAH in rats and the porcine pulmonary vein banding model of PAH [29]. In patients with PAH pulmonary arterial aldosterone levels are increased by 4.9-fold compared with controls [30]. In that study, aldosterone levels correlated inversely with cardiac output and positively with key measures of pulmonary vascular remodeling, including pulmonary vascular resistance and transpulmonary gradient. Adrenal stimulation by renin-angiotensin axis upregulation is likely to be the chief source of aldosterone production in PAH; however, the possibility that pulmonary vascular tissue functions as an extra-adrenal source of aldosterone production has been reported. Endocrine functionality was suggested originally five decades ago by several groups based on observations demonstrating that isolated synthesis of angiotensin I occurs in lung tissue [31]. In support of this are data demonstrating a transpulmonary increase in plasma levels of the aldosterone secretagogues angiotensin II [32] and the endothelin-1 precursor Big endothelin-1 [33] in patients with selected forms of pulmonary hypertension. Whether autonomous aldosterone synthesis in lung or pulmonary vascular tissue occurs or is relevant to PAH pathobiology in patients per se, requires further investigations [34].
It is noteworthy that key proteins required for de novo aldosterone synthesis, including steroidogenic acute regulatory protein (StAR), CYP11B1 (11-beta-hydroxylase), and CYP11B2 (aldosterone synthase) are constitutively expressed or inducible in cardiomyocytes, PASMCs, and/or PAECs by factors associated with pulmonary vascular injury [35, 36]. Treatment of human PAECs with ET-1 at levels similar to those observed in PAH patients induces association of the steroidogenic transcription factors steroidogenic factor-1 (SF-1) and PGC-1α to the CYP11B2 promoter, which increases pulmonary endothelial CYP11B2 expression and aldosterone levels by twofold in vitro (Fig. 9.1) [37]. Furthermore, pharmacological antagonism of the mineralocorticoid receptor with spironolactone attenuates angiotensin-II-mediated PASMC hypertrophy [35], providing some evidence in support of functionally active aldosterone biosynthesized in pulmonary vascular tissue.
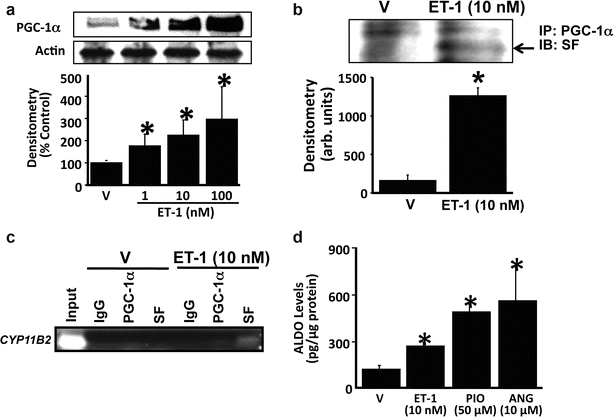
Fig. 9.1




Endothelin-1 (ET-1) stimulates PGC-1α-dependent association of steroidogenesis factor (SF) with CYP11B2 to increase aldosterone levels. (a) The effect of ET-1 on PGC-1α expression was assessed by Western analysis (n = 4). (b) Co-immunoprecipitation experiments demonstrated that incubation of human pulmonary artery endothelial cells with ET-1 (10 nM) for 24 h induced the association of PGC-1α with steroidogenesis factor-1 (SF) (n = 3). (c) Chromatin immunoprecipitation (n = 3) of cell lysates using antibodies to PGC-1α, SF, and immunoglobulin-G (IgG) as a negative control was followed by PCR amplification of the proximal region of the CYP11B2 (aldosterone synthase) promoter region containing the gonadotrope-specific element. (d) The functional effect of PGC-1α stimulation on aldosterone production was assessed in cells treated with the selective PGC-1α agonist pioglitazone (50 μM) for 24 h (n = 4), or with ET-1 (10 nM) or angiotensin II (ANG) (10 μM) for 24 h as positive controls. *p < 0.05 vs. V. PGC-1α PPAR-γ co-activator-1α; arb. units arbitrary units, IP immunoprecipitation, IB immunoblot (Data are presented as mean ± S.E.M. Representative blots are shown. Reproduced with permission from Maron et al. [37])
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
