DEFINITIONS
Acute kidney injury (AKI), previously called acute renal failure, is a common problem in the cardiac care unit (CCU) that is associated with increased morbidity and has an independent effect on the risk of short-term mortality. Traditionally, AKI has been defined as a rapid (i.e., over hours to weeks) and usually reversible decline in glomerular filtration rate (GFR). In an effort to standardize this subjective definition, numerous attempts have been made to define AKI more objectively. The most commonly accepted definitions are the RIFLE classification
1 and the acute kidney injury network (AKIN) criteria
2 that are listed in
Table 30.1.
A new terminology,
cardiorenal syndrome,
3 has recently been proposed to define the spectrum of disorders seen because of the hemodynamic interdependence between the heart and the kidneys. It includes those disorders of the heart and kidneys where acute or chronic dysfunction in one organ may induce acute or chronic dysfunction of the other. Five subtypes have been defined:
Type 1 acute cardiorenal syndrome: Acute worsening of cardiac function leading to renal dysfunction
Type 2 chronic cardiorenal syndrome: Chronic abnormalities in cardiac function leading to renal dysfunction
Type 3 acute renocardiac syndrome: Acute worsening of renal function causing cardiac dysfunction
Type 4 chronic renocardiac syndrome: Chronic abnormalities in renal function leading to cardiac disease
Type 5 secondary cardiorenal syndrome: Systemic conditions causing simultaneous dysfunction of the heart and kidney
EPIDEMIOLOGY AND RISK FACTORS
The incidence of AKI varies, with an average incidence of 5% in hospitalized patients and 25% in the intensive care unit.
4,
5 and
6 The incidence of AKI requiring renal replacement therapy (RRT) was 4% in a large, multicenter study.
5 The incidence varies significantly owing to the lack of standardized definitions used in the literature. In a large, tertiary center CCU, characteristics such as older age, African American race, diabetes, hypertension, previous coronary disease, and heart failure were incrementally more common across increasing renal dysfunction strata.
7 Decompensated heart failure, shock, use of iodinated contrast media, aggressive diuresis, and use of nephrotoxic medications are common precipitants of AKI.
PATHOPHYSIOLOGY AND APPROACH TO ACUTE KIDNEY INJURY
The kidney plays a key role in maintenance of fluid balance and excretion of waste products. Volume depletion is initially compensated by various systemic mechanisms in an attempt to restore tissue perfusion. Hypovolemia, for any reason, reduces release of atrial natriuretic peptide, increases production of antidiuretic hormone (ADH), and increases sympathetic (and decreases parasympathetic) activity through the baroreceptor reflex. In the kidney, the combination of hypotension and sympathetic activation leads to reduced perfusion pressure in the afferent arteriole and a decrease in the GFR. A decrease in tubular sodium chloride
(due to slower transit and increased proximal reabsorption) is sensed by the macula densa in the distal convoluted tubule, and this causes the juxtaglomerular apparatus to release renin. Renin activates angiotensin, which is converted peripherally to angiotensin II. Apart from being a potent vasoconstrictor, it also acts on the adrenal cortex to produce aldosterone, which increases salt and water reabsorption in the kidney. In addition to these mechanisms, the kidney possesses the ability to autoregulate, that is, maintain its perfusion pressure despite changes in mean blood pressure over a wide range of 80 to 180 mm Hg.
As a consequence of this compensatory mechanism, the renal tubules remain intact despite renal hypoperfusion. When normal renal perfusion is restored, urine flow returns to normal.
A concept that is important in understanding the pathophysiology of AKI in the CCU is that of effective circulating volume (ECV). ECV is the arterial blood volume effectively perfusing tissue. ECV is a dynamic quantity and not a measurable distinct compartment that normally varies directly with extracellular fluid (ECF). The ECV may not vary directly with the extravascular fluid volume in certain diseases, such as congestive heart failure (CHF) or hepatic cirrhosis; therefore, in these clinical settings, renal hypoperfusion may occur despite apparent volume excess.
In contrast to prerenal AKI, if the hypovolemia is severe or sustained owing to other toxic injuries, the renal tubules can become necrotic and lose their ability to conserve salt and water. Tubular obstruction by necrotic cells at the pars recta (where the proximal tubule narrows into the descending loop of Henle) leads to rise in the intraluminal pressure, decreasing the glomerular tubular gradient. This reduction in GFR may persist even after restoration of normal hemodynamics. Furthermore, injury to the tubular basement membrane can result in back leak of tubular fluid into the interstitial tissue. In this situation, urine flow may not be restored despite restoration of normal renal perfusion.
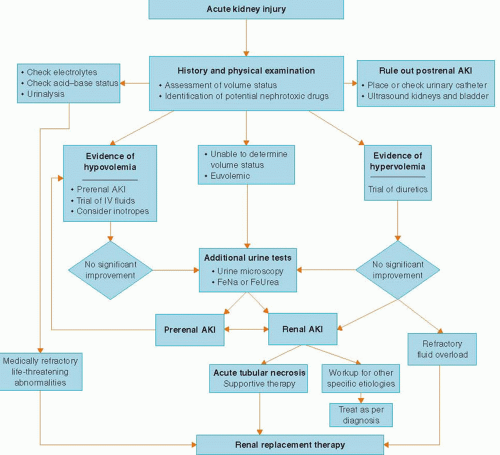
Based on this pathophysiology of renal adaptation to injury, the approach to AKI is the same in every clinical setting: delineation into the three pathophysiological categories of prerenal, intrinsic renal, or postrenal AKI (
Table 30.2). Prerenal azotemia results from ineffective renal perfusion and is the commonest cause of AKI in patients presenting to the emergency room. Renal or intrinsic AKI is secondary to an intrinsic renal disease and in the CCU most commonly results from ischemic or toxic acute tubular necrosis (ATN). The prerenal and intrinsic renal categories are not mutually exclusive but rather represent a continuum of renal injury where persistence of the ischemic insult may deteriorate into ATN. A prerenal state also sensitizes the kidney to nephrotoxic insults thus accounting for the increase in the incidence of AKI owing to nephrotoxic agents in hypovolemic states. Postrenal AKI owing to obstruction in the urinary collecting system is an uncommon cause of AKI in the CCU but should always be ruled out promptly because of its reversibility.
CLINICAL EVALUATION
Either a rise in the blood urine nitrogen (BUN) and serum creatinine or a drop in the urine output should alert the clinician to the presence of AKI. One of the earliest signs of AKI may be oliguria that is conventionally defined as urine output <400 ml per day. In the CCU, oliguria means insufficient urinary output for that particular patient, usually <0.5 ml/kg/hour. However, a patient who is fluid overloaded or with a high catabolic rate may require a urine flow rate considerably higher than this minimum standard. A number of etiologies of AKI present with nonoliguric rises in the BUN and creatinine; therefore absence of oliguria does not preclude an evaluation of renal injury.
The history should be focused on identification of risk factors for AKI, determination of baseline renal function to ascertain presence of chronic kidney disease (CKD) if possible, careful assessment of symptoms and precipitants of volume depletion (careful review of the intake and output), careful review of vital signs to identify episodes of hypotension, and a
thorough review of all home medications as well as all recently administered in-hospital medications to identify possible nephrotoxins. The history is essential in determining the etiology of AKI as it may lead the clinician to prevent further exposure to ongoing nephrotoxic insults.
Another important historical feature is the temporal correlation between the possible nephrotoxic insult and the clinical course of AKI. The serum creatinine rises rapidly (within 24 to 48 hours) in patients with AKI following renal ischemia and radiocontrast exposure. Peak serum creatinine concentrations are usually seen after 3 to 5 days with contrast nephropathy and return to baseline after 5 to 7 days. In contrast, serum creatinine concentrations usually peak later (7 to 10 days) in ATN and sometimes even later in atheroembolic disease. The rise in serum creatinine seen with many tubular epithelial cell toxins (e.g., aminoglycosides) or in the setting of drug-induced acute interstitial nephritis usually happens 7 to 10 days after onset of therapy.
“Information from the physical examination is invaluable and must be integrated with the historical and laboratory data. The patient’s volume status must be carefully assessed by examination of skin turgor, edema, jugular venous distension, orthostatic hypotension, and pulmonary vascular congestion. In addition, certain findings on physical examination can be diagnostic, for example, the presence of a distended bladder in a patient with urinary retention, a skin rash in suspected drug-induced interstitial nephritis, papilledema in malignant hypertension, or retinal cholesterol plaques in a patient with atheroembolic disease.