Fig. 9.1
Mechanisms involved in renal congestion in heart failure
How Does Renal Congestion Lead to Worsening of Renal Function?
As suggested above venous congestion (including renal vein) is an important factor for kidney dysfunction. However, little is known regarding the mechanisms leading to deterioration of renal function in venous congestion. Some mechanisms such as reduced transglomerular pressure, increased interstitial pressure, interstitial fibrosis, tubular back leak probably play a role. Additionally, increased sympathetic renal nerve activity resulting in intrarenal arterial vasoconstriction and a fall in GFR play a role [25]. During renal venous hypertension there would be a rise in renal interstitial pressure that would affect the entire capillary bed and the tubules, possibly also involving local hypoxia. Compression of the tubules raises the luminal pressure, further attenuates the transglomerular pressure gradient, and lowers the GFR. It is important to appreciate that a rise in renal interstitial pressure due to venous congestion is physiologically different than that caused by elevations in arterial pressure which is associated with a natriuresis [26].
The inflammatory process is another mechanism which is thought to be involved in venous congestion mediated deterioration of kidney function [27, 28]. Inflammation can beget vascular dysfunction via endothelial activation and enhanced arterial stiffness. Second, inflammation may reduce myocardial contractility either through functional suppression of the contractile apparatus or through increased myocardial cell death. Third, inflammation may cause progressive renal dysfunction and fibrosis. Finally, inflammation may increase the permeability of the endothelium allowing extravasation of fluids into the alveolar space of the lungs and absorption of pro-inflammatory endotoxin from the bowel [27]. In heart failure and venous congestion activation of renin angiotensin system (RAS) and sympathetic system promotes inflammatory reaction. However, accumulating evidence suggests that volume overload and venous congestion independent of RAS and sympathetic system, are independently accepted as an additional source of inflammatory mediators [27]. Thus evidence is accumulating that inflammation is related with congestion. However, the exact mechanisms related to venous congestion and inflammation are not solved completely although some mechanisms are speculated. In volume overload state mesenteric venous congestion leads to bowel wall oedema with translocation of gram-negative bacteria through the endothelial cells of the intestinal villi. Lipopolysaccharide is then released into the circulation, and activating the inflammatory response [28]. The proof of this assumption comes from recent studies [29, 30]. Similarly, in a recent prospective study, endotoxin levels were higher in chronic kidney disease (CKD) patients with signs of fluid overload compared to CKD patients without fluid overload [31].
The other speculated mechanism is the activation of endothelial cells during congestion. Venous congestion itself may switch the synthetic and endocrine profile of the endothelium from quiescent toward an activated state that is pro-oxidant, proinflammatory, and vasoconstricting. Once “activated,” the endothelium can promote additional congestion through humoral, renal, and cardiac mechanisms, resulting in a deleterious positive feedback loop that leads, over time, more congestion [19].
Indeed it was clearly shown that during venous stretch endothelin-1 (ET-1), interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-alpha) can be secreted within hours of stretch exposure [32]. Besides, biomechanical signals such as stretch modulate endothelial production of reactive oxygen species [33]. Reactive oxygen species and cytokines may also trigger an inflammatory response through activation of nuclear factor (NF)-κB [34].
In summary, based on these reports, vascular stretch can activate endothelial pro-oxidant and proinflammatory programs. These results demonstrate that venous congestion and volume overload alone can promote an inflammatory state with elevations of inflammatory mediators in the circulation. Ultimately, the source of chronic inflammation in venous congestion syndrome is likely a combination of the several biologic mechanisms discussed (Fig. 9.2).
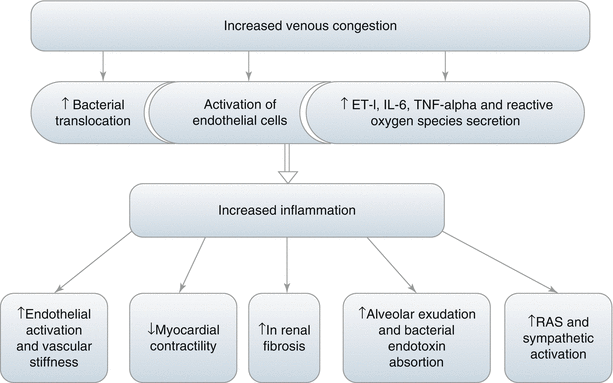
Fig. 9.2
Mechanisms of increased inflammation during venous congestion
Venous Congestion: An Important Cause of Renal Dysfunction in Heart Failure
Various studies have shown that venous and renal congestion may play more important role in kidney dysfunction. In the ESCAPE (Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness) trial involved hospitalized decompensated heart failure patients in which kidney function did not correlate with cardiac index, pulmonary capillary wedge pressure, or systemic vascular resistance, but rather was associated with right atrial pressure [35]. In a retrospective analysis of 2,557 patients undergoing right heart catheterization, CVP was associated with low estimated GFR independently from cardiac index, and it predicted mortality [23]. Similarly, Guglin et al. described catheterization findings in 178 heart failure patients wherein low estimated GFR correlated with high CVP and low renal perfusion pressure but not the cardiac index or left ventricular ejection fraction [36]. Aranson et al. studied 475 patients with decompensated heart failure, of which 238 had right heart catheterization data available over the first 24 h. Net fluid loss was recorded in the first 24 h. Worsening renal function was defined as a >0.3 mg/dL increase in serum creatinine above baseline. The authors found that baseline right atrial pressure had a weak association with baseline renal function (r: −0.17, p: 0.009), but there was no association between baseline or change in right atrial pressure with the volume removed over the first 24 h, nor was there an association with the incidence of worsening renal function up to 14 days. The authors did, however, find a strong association between an increased volume of diuresis in the first 24 h of the hospitalization and a lower incidence of worsening renal function. The authors concluded that early net fluid loss is associated with worsening renal function [37]. Thus, as detailed below CVP may not be sensitive enough to detect subtle changes in volume status.
The role of haematocrit elevation (as a measure of extracellular fluid reduction) was also investigated in heart failure patients. In one study it was shown that 1,684 patients with heart failure were compared with respect to all cause mortality, cardiovascular mortality or heart failure depending on whether haemoconcentration (as a marker of decongestion) occurred or not. Haemoconcentration was defined as ≥3 % absolute increase in haematocrit. Haemoconcentration correlated with greater risk of in-hospital worsening renal function, but renal parameters generally returned to baseline within 4 weeks post-discharge. Patients with haemoconcentration were less likely to have clinical congestion at discharge and experienced greater in-hospital decreases in body weight and natriuretic peptide levels. They were also less likely to have dyspnoea, rales, and peripheral oedema at the time of discharge/day 7. After a median follow-up of 9.9 months and after adjustment for baseline clinical risk factors, every 5 % increase of in-hospital haematocrit change was associated with a decreased risk of all-cause death [hazard ratio (HR) 0.81, 95 % confidence interval (CI) 0.70–0.95]. Haematocrit change was also associated with decreased cardiovascular mortality or heart failure hospitalization at ≤100 day’s post-randomization (HR 0.73, 95% CI: 0.71–0.76). The authors concluded that haemoconcentration was associated with greater improvements in congestion and decreased mortality and heart failure re-hospitalization despite an increased risk of in-hospital worsening renal function in heart failure patients [38]. In the PROTECT trial authors also found haemoconcentration, defined as absolute in-hospital increases in haemoglobin, to correlate with favourable prognosis despite a decrease in renal function [39]. In the Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness (ESCAPE) trial patients with haemoconcentration experienced greater net weight loss and substantially lower risk of mortality despite increased risk for worsening renal function [40]. On the other hand, Davila et al. demonstrated that in-hospital increases in haemoglobin associated with worsening renal function, but not mortality [41]. At this point one must mention that anaemia and iron deficiency may be important covariates in heart failure. As well known anaemia and iron deficiency is common in CKD. However, recent evidence suggests that these parameters are also important in heart failure. Indeed, patients with heart failure may be prone to the development of iron deficiency as a consequence of a depletion of iron stores or defective iron absorption and the reduced availability of iron recycled in the reticuloendothelial system [42, 43]. Indeed, in one prospective study it was shown that Treatment with ferric carboxymaltose for 24 weeks in patients who had chronic heart failure and iron deficiency with or without anaemia improved symptoms, functional capacity, and the quality of life. Importantly this benefit was seen in all patients with and without anaemia [44]. Thus apart from its role in showing extracellular fluid reduction, anaemia and iron deficiency may play a role in worse outcomes in heart failure patients with heart failure.
Is Renal Function Deterioration During Heart Failure Treatment Good or Bad?
Classically, it was accepted that deterioration in renal function was associated with increased mortality among patients with heart failure [45, 46]. A retrospective analysis of the ADHERE database suggests that serum creatinine >2.75 mg/dl is a significant risk factor for mortality in patients with heart failure [47]. However, recent findings challenged this concept and showed that deterioration in renal function had either no effect or beneficial impact [35, 48–52]. The cause of these contrasting findings is unknown but the effects of congestion may be responsible. To address this issue the independent effect of deterioration in renal function and presence of congestion during discharge in acute heart failure patients were investigated by Metra et al. Congestion was defined as the persistence of one or more signs or symptoms of fluid overload at discharge. The following symptoms and signs were prospectively considered: third heart sound, pulmonary rales, jugular venous stasis, hepatomegaly, and peripheral oedema. The outcome measures were post discharge mortality and acute heart failure readmission. There was no difference with respect to outcomes in patients with deterioration in renal function and no congestion and without deterioration in renal function and no congestion. However, the outcomes were worse in patients with congestion alone (without deterioration in renal function) and with congestion and deterioration in renal function. In the last patient group the hazard ratio for mortality was 2.44 (CI: 1.24–4.18). The authors concluded that deterioration in renal function alone, when detected using serial serum creatinine measurements, is not an independent determinant of outcomes in patients with acute heart failure. It has an additive prognostic value only when it occurs in patients with persistent signs of congestion [53]. Taken together, these data suggest that change in congestion status is perhaps a key variable underlying the impact of deterioration in renal function in this population.
Second selection bias may be another explanation for contrasting findings. For example, sicker patients who are more congested and have a longer hospital stay tend to have more creatinine measurements done and hence have a greater likelihood of showing a creatinine increase. Thus an increase in serum creatinine would be simply a marker of more severe heart failure rather than of progressive kidney disease [53]. Third, increases in serum creatinine levels may just be caused by renal haemodynamic abnormalities and diuretic therapy [5]. Indeed, low cardiac output and increased CVP and renal vein pressure may cause a reduction in the glomerular filtration pressure, progressive kidney disease and resistance to furosemide administration [23, 24]. Fourth, CVP a common surrogate for venous congestion was recently found to track poorly with fluid removal [37, 54]. The highly compliant nature of the venous system enables large changes in blood volume to be associated with small changes in pressure. Thus, even an effective treatment of volume overload may not be sufficient to produce a meaningful reduction in CVP and, in turn, reduce the risk of renal impairment. Besides, in most of the experimental studies showing relationship with venous pressure and deterioration in kidney function, the venous pressure was abruptly raised to extremely high values that are usually not seen even in patients with severe heart failure (e.g. 25–50 mmHg) [55]. For example, in the isolated perfused rat kidney model, GFR was not significantly altered until the imposed venous pressure reached 25 mmHg [56]. However, it is also possible that the kidneys may be more sensitive to elevated CVP in the setting of heart failure, such that GFR may fall with moderate CVP elevations [57]. Thus all these issues may be responsible for these divergent results. Indeed, today we still don’t know why some decompensated heart failure patients exhibit improvements in renal function with diuresis, whereas others display renal function deterioration, limiting attainment of euvolemia. Based on current data, one can speculate that deterioration in kidney function during heart failure is an adverse event given the fact that if the insult is progressive and long-lasting. However, transient deterioration in renal function may not pose worse prognosis.
Markers for Worsening Renal Function During Heart Failure
Although the detailed description of markers used to detect renal damage during heart failure is beyond the scope of this chapter some important points must be remembered.
BUN and Creatinine
Blood Urea Nitrogen (BUN) is one of the most used and traditional markers for detecting renal function. A pooled analysis of the Prospective Randomized Evaluation of Cardiac Ectopy with Dobutamine or Nesiritide Therapy (PRECEDENT) trial conducted in 541 New York Heart Association (NYHA) Class III-IV, heart failure patients with systolic dysfunction assessed the prognostic importance of four different measures of renal function namely BUN, serum creatinine, BUN/creatinine ratio and estimated creatinine clearance. After 1-year follow-up, BUN was the only significant predictor of mortality, with an adjusted relative risk (RR) of 2.3 in patients in the upper compared with the lower quartiles (95 % CI 1.3–4.1; p: 0.005). BUN/creatinine ratio yielded similar prognostic information as BUN (adjusted RR = 2.3; 95 % CI 1.4–3.8; p: 0.007) for patients in the upper compared with the lower quartiles [58]. Consistently, a post hoc analysis of the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Chronic Heart Failure (ACTIV in congestive heart failure) trial showed that among 319 patients with reduced systolic function stratified into quartiles according to baseline BUN, those in the highest quartile (40 mg/dL) had the highest 60-day mortality compared with those in the lower quartile (14.3 vs. 0 % respectively, p: 0.001) and the highest rate of death or heart failure hospitalization (30.0 vs. 8.6 %, p: 0.001). After adjustment for covariates, BUN remained a significant predictor of both mortality and the composite endpoint of death or heart failure hospitalization at 60 days after hospital discharge. Serum creatinine and creatinine clearance did not predict mortality after covariate adjustment [59]. Other studies also show its relation with morbidity and mortality in patients with heart failure [58, 59]. In the randomized Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF), BUN had a stronger relationship with outcomes as compared with GFR, calculated on the basis of serum creatinine levels [60]. Thus there is growing evidence that urea is a stronger predictor of outcome than creatinine in patients with heart failure. The exact cause of this association is not known but BUN has been accepted as a mediator of neuro-humoral activation during heart failure. Additionally, drawbacks of serum creatinine may play a role. These include the dependence of serum creatinine to other variables, namely, age, gender and muscle mass, the insensitivity of serum creatinine in detecting early renal injury (relatively large amount of renal damage can occur without producing a change in GFR calculated on serum creatinine levels which start to increase only at advanced stages of renal dysfunction) and inability of serum creatinine to measure renal injury. For example changes in renal function may occur as a consequence of changes in volume status in the absence of any renal damage.
Cystatin C
It has been suggested that cystatin C might be superior to creatinine in terms of predicting prognosis in patients with heart failure [61–63]. Wen et al. explored early markers of renal impairment in experimental post-myocardial infarction heart failure and found that it is the high blood cystatin C levels, rather than serum creatinine and BUN that predict increased post-MI heart failure incidence [64]. In acute heart failure, some studies have demonstrated that cystatin C can be a good prognostic marker. Lassus et al. measured cystatin C on admission and at 48 h in 292 patients hospitalized for acute heart failure. Acute kidney injury defined by an increase in cystatin C 0.3 mg/l within 48 h. The increase of cystatin C occurred in 16 % of patients. This increase was associated with longer length of hospitalization (P: 0.01) and with a significantly higher in-hospital mortality (odds ratio 4.0 95 % CI 1.3–11.7, P: 0.01). At 90 days, the increase in cystatin C was an independent predictor of mortality (adjusted odds ratio 2.8 95 % CI 1.2–6.7, P: 0.02) [65]. However, given the fact that studies regarding the cystatin in heart failure are scarce, more studies are needed to explore the role of cystatin C in patients with heart failure.
Neutrophil Gelatinase-Associated Lipocalin (NGAL)
In patients with acute heart failure, neutrophil gelatinase-associated lipocalin (NGAL) can predict deterioration of kidney function more accurately and in an earlier stage than serum creatinine. In fact, NGAL level rises about 24 h before serum creatinine values. In 91 patients admitted for acute heart failure, deterioration of kidney function was observed in 38 % within 5 days of follow-up. Patients who developed deterioration of kidney function had significantly higher median admission serum NGAL levels (194 ng/ml vs. 128 ng/ml, P = 0.001) with an increase in risk of developing deterioration of kidney function [66]. Besides, it was suggested that levels of urinary NGAL may be more sensitive as makers of tubular damage than serum levels [67, 68].
Kidney Injury Molecule 1 (KIM-1)
In chronic heart failure, Kidney injury molecule 1 (KIM-1) demonstrated a correlation with plasma N-terminal pro-brain natriuretic peptide levels and, independently of GFR values; it was associated with an increased risk of death or heart failure hospitalizations [68]. KIM-1 is highly sensitive to acute tubular injury but in the setting of acute heart failure its role is still unsettled.
Uric Acid
Current evidence suggests that uric acid may be either a marker of poor prognosis [69] or an active player in the pathogenesis of heart failure [70]. A study of 112 NYHA class III or IV patients found that serum uric acid level is a strong predictor of poorer outcomes, defined as mortality and need for transplant [71]. Janakowska et al. had similar findings regarding elevated uric acid levels and poorer outcomes in 119 NYHA class I–III patients, suggesting uric acid levels correlate with mortality and morbidity even in mild heart failure [72]. The Framingham Offspring cohort showed that incidence rates of heart failure were approximately sixfold higher among those in the highest quartile of uric acid compared with those at the lowest quartile [73]. The relationship between uric acid and heart failure was thought to be associated with inflammation, renal congestion and oxidative stress. The mechanism of renal congestion is a special concern. In 50 patients with reduced left ventricular systolic function, uric acid correlated significantly with pulmonary artery pressure, pulmonary capillary wedge pressure, and with clinical signs of volume overload (rales, oedema, and paroxysmal nocturnal dyspnoea). It also inversely correlated with left ventricular ejection fraction, suggesting that uric acid may be a non-invasive indicator of elevated filling pressures [74]. These findings were further supported by Kittelson et al. who found that not only were higher uric acid levels were associated with increased pulmonary artery and wedge pressure but with increased right atrial pressures as well [75]. Moreover, when patients with heart failure were monitored longitudinally, uric acid correlated with clinical status of patients. Odds ratios of hyperuricemia were 1.67 (95 % CI, 1.21–2.32) for heart failure decompensation and 0.21 (95 % CI, 0.08–0.55) for compensation [76].
Natriuretic Peptides
It was stated that patients admitted with acute breathlessness due to heart failure and an elevated natriuretic peptide level (generally 600 pg/ml for B-type natriuretic peptide (BNP) or 6,000 pg/ml for N-terminal pro-BNP) have a high filling pressure secondary to volume overload [77]. High atrial natriuretic peptide (ANP) levels not only would be a marker of venous congestion, but there is also evidence that its beneficial natriuretic effects are attenuated in heart failure. High renal interstitial pressure due to venous congestion may impair preservation of GFR by loss of ANP’s effects modulating transforming growth factor β [78]. A recent study has shown that BNP levels correlate with capillary wedge pressure, it can also serve as an indirect marker for deterioration of kidney function during the treatment of acute decompensated heart failure [79].
Vasopressin
Excess vasopressin levels have long been recognized in patients with heart failure, particularly those with severe clinical manifestations and have the potential to exert deleterious effects on various physiological processes in heart failure [80]. However, measurement of circulating vasopressin levels has been challenging because it is released in a pulsatile pattern, unstable and is rapidly cleared from plasma. Arginine vasopressin is derived from a larger precursor peptide (preprovasopressin) along with copeptin, which is released from the posterior pituitary in an equimolar ratio to arginine vasopressin and is more stable in the circulation and closely reflects arginine vasopressin levels. Copeptin levels have been found to closely mirror the production of arginine vasopressin and have been proposed as a prognostic marker in acute illness [81]. Thus studies are needed whether vasopression and/or copeptin levels are promising in heart failure.
Bioimpedance
Bioimpedance technique was introduced to study changes in the volume in a tissue in 1940s. Bioimpedance is based on the principle that fluid (oedema) is a good conductor of electrical current and is associated with a low impedance value. Decreased bioimpedance reflects total body water excess, with total body water derived from these values by making certain electrophysical assumptions [82]. Bioimpedance vector analysis has been used in heart failure patients [83, 84]. More studies are needed regarding the efficiency of bioimpedance in heart failure patients.
Treatment of Renal Congestion in Heart Failure
There are various treatment options in renal congestion in heart failure (Table 9.1). The main aim is the decongestion of excess fluid without compromising renal function. The forthcoming section deals with the main treatment strategies regarding decongestion in heart failure.
Table 9.1
Treatment strategies of venous congestion in heart failure
Loop diuretics | Use of intravenous route is more effective |
Bolus dosing may be as affective as continuous infusion [48] | |
Start the initial dose at 2–2.5 times the ordinary oral dose [48] | |
Increase dose until the adequate symptom relief is achieved – in case of hypotension consider continuous infusion | |
Multiple dosing more effective than single dosing | |
Consider adding low-dose thiazide diuretics in case of resistance; monitor electrolytes carefully | |
Ultrafiltration | Peripheral veno-venous ultrafiltration |
Peritoneal dialysis | |
V2R antagonists | |
Experimental evidence suggest improved survival [87] | |
Augment the diuretic and the natriuretic response to furosemide [88] | |
Adenosine receptor blockers | Dilatation of afferent arteriole and preservation of GFR |
Likely not causing worsened renal function [89] | |
Favorable effect on dyspnea and short-term mortality [89] | |
May be associated with higher rates of seizures and stroke [90] | |
Dopamine | |
In acute HF use with caution [93] | |
No clear effect on mortality, rehospitalizations and prevention of renal damage [94] | |
Compared with placebo, no effect of low-dose dopamine on decongestion, renal function, or clinical outcomes [95] | |
Natriuretic peptides | Reduce cardiac filling pressure, increase cardiac output, promote diuresis and reduce RAS and release of norepinephrine [96] |
Borderline effect on dyspnea [97] | |
May have hypotensive effect | |
Compared with placebo, no effect of nesiritide (recombinant BNP) on decongestion, renal function, or clinical outcomes [95] | |
Novel therapies | Spliced BNPs potential effects [98] |
Hypertonic saline with furosemide [99] | |
Relaxin [100] |
Loops Diuretics
Loop diuretics are commonly used in patients with heart failure. The detailed action of loop diuretic is beyond the scope of this chapter however some important issues must be remembered. First of all, in heart failure, the dose-response curve shifts downward and to the right and there is possibility that drug reaching the Henle’s loop decreases which necessitates a higher dose to achieve the same effect. Thus reaching a therapeutic threshold is an important concept [101]. At this point on may argue that higher loop diuretic dosing in heart failure is associated with worse clinical outcome [102]. However, these studies are criticized because patients with more severe disease and underlying renal insufficiency require higher diuretic doses [48]. This issue is examined in a recent trial which 183 patients with advanced heart failure stratified by baseline diuretic dose (furosemide < 80 or > 80 mg daily). Patients receiving high-dose diuretics (n = 113) had more markers of increased cardiovascular risk and were more likely to have had a recent history of clinical instability (33 % vs. 4 %). After adjusting for clinical stability, diuretic dose was no longer a significant predictor of increased risk [103]. Thus it is possible that a subgroup of patients with heart failure refractory to diuretics and thereby requiring higher doses of these drugs during heart failure decompensation are especially susceptible to the development of renal injury. In fact, these patients had significantly higher admission serum creatinine levels and included a higher percentage of patients requiring thiazide in addition to loop diuretics. Alternatively, patients with deterioration of kidney function required higher diuretic doses because of more advanced heart failure. Thus, the question of whether higher diuretic doses are responsible for deterioration of kidney function or are a marker of greater disease severity remains unanswered [104]. Secondly, in general loop diuretics are given as a single daily dose. However, a single furosemide bolus especially in heart failure, elicits a transient period of natriuresis followed by a longer period of increased renal tubular sodium avidity due to increase in systemic vascular resistance, plasma renin activity, and plasma levels of norepinephrine and arginine vasopressin [105, 106]. Thus there is an escape from the action of loop diuretics especially when given as a single dose. To overcome this effect, loop diuretic can be administered two or more times per day. Indeed, continuous loop diuretic infusion is an extrapolation of this concept in which the time interval between dosing is reduced to zero. In this scenario, there is no antinatriuretic rebound period and negative sodium balance is sustained [4]. However, it was shown that continuous dosing is not more effective than an intelligently prescribed bolus regimen as proven in the Diuretic Optimization Strategies Evaluation (DOSE) trial [48]. However, infusion may be preferred in patients with low systolic blood pressure. Thirdly, to overcome this escape the addition of a non-loop diuretic (i.e., thiazide or potassium-sparing diuretic) may be effective by decreasing the enhanced sodium absorption in the distal tubule above [107]. The excessive use of diuretics and sympathetic overactivity in heart failure promotes the activity of the RAS. Activation of the RAS leads to excessive salt and water retention and vasoconstriction of the venous beds, altering cardiac preload and afterload, which further worsens renal function. Thus addition of mineralocorticoid receptor antagonists, such as spironolactone and eplerenone, may attenuate the neurohumoral surge and prevent deterioration of kidney function. Previous small, nonrandomized, open-label trials have shown that these drugs at high doses overcome diuretic resistance in heart failure without a significant effect on serum creatinine [108, 109]. However, in an acute setting in which aggressive diuresis is needed, these drugs may also cause deterioration of kidney function. The balance is tight and careful attention to renal parameters should be given in such patients, with adjustment of doses of both drugs. Sometimes, it is prudent to withhold ACE inhibitors and angiotensin receptor blockers particularly in patients at high risk of developing renal injury, such as patients with advanced age and aggressive diuresis. Lastly, loop diuretics may also have independent actions of tubules irrespective of systemic and renal haemodynamic effects. In one interesting study in 30 patients with chronic systolic heart failure the effect of loop diuretic withdrawal and reinitiation on tubular dysfunction was evaluated. At baseline, subjects were withdrawn from their loop diuretics. After 72 h, their furosemide regimen was reinstated and patients were studied again 3 days later. Serum creatinine, atrial and B-type natriuretic peptide, KIM-1, urinary N-acetyl-beta-D- glucosaminidase (NAG), and serum as well as urinary NGAL were determined at various time points. Diuretic withdrawal resulted in increases in atrial and B-type natriuretic peptide (both p < 0.05). Serum creatinine was unaffected. Both urinary KIM-1 (p < 0.001) and NAG (p < 0.010) concentrations rose significantly, after diuretic withdrawal, whereas serum and urinary NGAL were not significantly affected. After reinitiation of furosemide, both urinary KIM-1 and NAG concentrations returned to baseline (both p < 0.05), but NGAL values were unaffected. The authors concluded that subclinical changes in volume status by diuretic withdrawal and reinstitution are associated with increases and decreases of markers of tubular dysfunction in stable heart failure and diuretic therapy may favourably affect renal and tubular function by decreasing congestion [110].
At this point one must also mention the importance of salt and fluid restriction in heart failure. Although salt and fluid restriction is the primary dietary therapy heart failure, this is mostly based on expert opinion and few controlled studies are available. Albert et al. demonstrated that fluid restriction (1,000 mL/day) in hyponatremic (serum sodium <137 mg/dl) patients improved quality of life at 60 days after discharge [111]. Hummel et al. showed that in heart failure patients with preserved ejection fraction sodium-restricted diet was associated with favourable changes in ventricular diastolic function, arterial elastance, and ventricular–arterial coupling [112].
Diuretics vs. Ultrafiltration to Relief Renal Congestion
Diuretic therapy aimed at fluid withdrawal and relief of congestion which are the main currently available strategies for reducing venous congestion in decompensated heart failure. Ultrafiltration is another option for the same purpose. Although several guidelines state that ultrafiltration is reasonable for patients with refractory congestion not responding to medical therapy [115–117]; we do not know known which of the two is safer and more effective [118, 119]. Although the two treatments seem to work for decongestion some differences must be mentioned. Firstly, the amount of urine produced in response to IV diuretics is not predictable but fluid removal by ultrafiltration is completely controllable and adjustable. Secondly, a potential advantage of ultrafiltration over loop diuretics is that the ultrafiltrate is isotonic, whereas the urinary output with loop diuretics is hypotonic therefore ultrafiltration removes more sodium (and less potassium) than diuretics for an equivalent volume loss [120]. Thirdly, if fluid removal does not exceed the interstitial fluid mobilization rate of approximately 15 ml/min, then the intravascular volume can be preserved with ultrafiltration, potentially interrupting the vicious cycle of neurohormonal activation and renal impairment that can occur with loop diuretics [121]. Besides, adequacy of intravascular re-fill during ultrafiltration can be assessed by continuous monitoring of the haematocrit with sensors placed in the withdrawal line. When the haematocrit does not significantly change during ultrafiltration, regardless of the amount of fluid removed, this indicates a proportional shift of water from the extravascular to the intravascular space. An increase in haematocrit may indicate either that plasma re-fill rate is inadequate or that interstitial oedema has been eliminated. This hypothesis is supported by data demonstrating that patients receiving ultrafiltration have lower plasma renin, norepinephrine and aldosterone levels as long as 90 days after treatment compared with those receiving diuretics [122]. Fourthly, elimination of proinflammatory cytokines [26, 123] or sodium-retaining vasoconstrictive agents may occur during ultrafiltration which is potentially involved in improvement in urinary output or restoration of diuretic responsiveness during ultrafiltration [121, 124]. Lastly, ultrafiltration mediated neurohumoral regulation is more sustained. This is probably a reason why improvement in clinical signs and symptoms of volume overload and functional capacity were found to be persistent [122, 125] and rehospitalisations are lower in ultrafiltration compared in to diuretic therapy [126]. However, as stated earlier there are no strict recommendations regarding the preferential use of diuretics, ultrafiltration and or combination. In fact in the Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) trial it was found that the use of a stepped pharmacologic-therapy algorithm was superior to a strategy of ultrafiltration for the preservation of renal function at 96 h, with a similar amount of weight loss with the two approaches Ultrafiltration was associated with a higher rate of adverse events [49].
Peritoneal Dialysis
In peritoneal dialysis, there is a continuous slow ultrafiltration leading to a reduction in fluid overload. Lowering afterload and lowering CVP could be important physiological mechanisms by which peritoneal dialysis leads to clinical improvement in heart failure symptoms. Moreover, it provides some replacement of renal function, by removing metabolic waste products, so a possible decrease in renal function will be better tolerated. However, the role of peritoneal dialysis in heart failure and renal congestion is not examined satisfactorily. Additionally, there are no studies available comparing peritoneal dialysis, diuretic therapy and salt restriction head-to-head in congestive heart failure [127]. In some studies, there were improvements to echocardiographic or other cardiac parameters with ultrafiltration by peritoneal dialysis [128–130]. There are also reports that peritoneal dialysis can reduce the number of hospital admissions and improve quality of life in chronic heart failure patients, as supported by recent non-randomized and non-controlled prospective studies [131–133]. However, the number of studied patients is limited and these trials all lack a control group. Also most studied patients had advanced renal failure contributing to fluid overload, introducing a major confounding factor. Another potential concern is the elevation of intra abdominal pressure by peritoneal dialysis. Currently, neither the optimal dialysis schedule nor the optimal fluid status to be reached in heart failure patients is known. The available reports all describe different peritoneal dialysis intervals, types, fluids and dosages.
Vasopressin Type 2 Receptor Antagonists
Recently, vasopressin type 2 receptor (V2R) antagonists have shown promise for use in patients with heart failure by increasing free-water excretion and serum sodium level [93]. For example, the oral V2R antagonist tolvaptan caused an early and sustained reduction in body weight and improvement in serum sodium in the EVEREST trial although did not improve mortality or morbidity [85, 86]. In a recent experimental study, chronic tolvaptan administration in a rat hypertensive heart failure model was examined considering the functional and pathological effects on both the myocardium and kidney. The animals were chronically treated with low-dose or high-dose (HD) tolvaptan or vehicle from the left ventricular (LV) hypertrophic stage. Chronic tolvaptan treatment persistently increased urine volume but did not affect blood pressure. In the HD group, the animal survival significantly improved (log-rank test, P < 0.01). At the heart failure stage, the progression of LV dysfunction was prevented and lung congestion was suppressed. Activation of atrial natriuretic peptide, endothelin-1, AVP, and V1aR mRNA levels were significantly suppressed in the LV myocardium. Meanwhile, renal histopathologic damage including tubular fibrosis and glomerulosclerosis was ameliorated and renal function was improved in the HD group at the heart failure stage. Concomitantly, not only activation of aquaporin-2 but also those of V2R, V1aR, renin, and endothelin-1 in the kidney were significantly suppressed (all P < 0.05). V2R antagonists also caused redistribution of AQP2 from apical to intracellular domains [87]. Goldsmith et al. studied the effect of conivaptan on renal and hormonal effects compared with or in combination with loop diuretics in stable heart failure patients. There were no significant effects of conivaptan, furosemide, or the combination on any haemodynamic variable, neurohormonal level, renal blood flow, or glomerular filtration rate. Conivaptan and furosemide similarly increased urine volumes; the effect of the combination was significantly greater. Furosemide, but not conivaptan, increased urinary sodium excretion, and the combination was significantly greater than after furosemide alone. Without adversely affecting important haemodynamic variables, neurohormones, renal blood flow, or glomerular filtration rate, conivaptan significantly augmented both the diuretic and the natriuretic response to furosemide in patients with chronic heart failure [88].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


