Remodeling of the Cardiac Interstitium in Heart Failure
Karl T. Weber
Yao Sun
Syamal K. Bhattacharya
Robert A. Ahokas
Ivan C. Gerling
Chronic cardiac failure (CCF) is a major health problem. Of the more than 4 million Americans already diagnosed with CCF, the great majority have atherosclerotic coronary artery disease with previous myocardial infarction (MI) (1,2). CCF appears over the course of several years in patients who develop progressive post-MI left ventricular dilatation in association with depressed resting ejection fraction (3,4,5,6). In others, ventricular enlargement post-MI appears early and is nonprogressive with preserved systolic performance. However, diastolic dysfunction may be evident during physical activity.
Ventricular diastolic and/or systolic dysfunction is related to a deterioration of previously normokinetic segments (i.e., sites remote to MI). Although loss of cardiac myocytes at and remote to the site of previous infarction(s) is believed to contribute to ventricular systolic dysfunction that appears in ischemic cardiomyopathy (ICM), despite hypertrophy of viable myocytes (7,8), an additional loss of myocytes through programmed cell death, or apoptosis, has recently been called into question (9,10,11,12). Abnormalities of viable myocyte contraction and relaxation have long been considered an additional cause of diastolic and systolic dysfunction in ICM (13,14). In addition to myocyte loss and faulty myocyte mechanics, a broader paradigm has recently emerged for the development of cardiac failure, one involving structural remodeling of noninfarcted myocardium by fibrous tissue (15,16,17).
Cardiac myocytes are highly specialized and highly differentiated parenchymal cells that impart cardiac tissue with unique morphological and functional features. Given their size, myocytes occupy 70% of the myocardium’s structural space. On the other hand, myocytes represent all but one-third of all cells found in the myocardium; the remaining two-thirds are nonmyocyte cells, such as fibroblasts. Fibroblasts are undifferentiated and have a pluripotent portfolio of responses, including marked phenotypic diversity and metabolic activity. They contribute to ICM by accounting for a progressive accumulation of fibrous tissue remote to MI, which can be independent of myocyte loss.
In the failing human heart with ICM, an infarct scar(s) is a common feature. Such replacement fibrosis rebuilds myocardium, restores structural integrity at sites of myocyte loss, and resists tissue deformation. Connective tissue also appears remote to previous infarction, where it presents as an interstitial fibrosis and microscopic scars. Not unlike other organs, where interstitial fibrosis represents a final common pathway to organ failure (18), it is this adverse accumulation of matrix in viable myocardium that is considered the major component to the adverse structural remodeling found in the explanted, failing human heart with ICM (16). The interstitial compartment in ICM is anything but acellular and inert. Herein we review the structural remodeling of the cardiac interstitium seen in ICM, together with molecular and cellular responses associated with such remodeling.
Normal Cardiac Interstitium
The extracellular space has multiple functions that make it an essential component of the myocardium. Its fibrillar collagen network, for example, serves to maintain tissue architecture and myocyte alignment throughout the cardiac cycle. It facilitates the transmission of myocyte-generated force to ventricular chambers (19). Additionally, the energy imparted to coiled collagen fibers during myocyte contraction contributes to myocyte relengthening during
relaxation and active early ventricular filling (20). Of equal importance, the interstitium contains tissue fluid through which oxygen, ions, and macromolecules reach myocytes and by which various metabolic end-products exit via venous and lymphatic capillaries. The normal cardiac interstitium is integral to myocyte contraction and nutrition and has been reviewed elsewhere (21).
relaxation and active early ventricular filling (20). Of equal importance, the interstitium contains tissue fluid through which oxygen, ions, and macromolecules reach myocytes and by which various metabolic end-products exit via venous and lymphatic capillaries. The normal cardiac interstitium is integral to myocyte contraction and nutrition and has been reviewed elsewhere (21).
Fibrillar Collagens
Type I and III fibrillar collagens are the major structural proteins of the extracellular matrix (22). They normally occupy 4% of its structural space (23). Type I collagen predominates and has the tensile strength of steel (24). Type III collagen is far less abundant but is more distensible. The fibrillar collagen network of the ventricles is a structural continuum with its exteriorized portion found in chordae tendineae and valve leaflets (25). The network is segregated into various components termed epimysium, perimysium, and endomysium (19,25,26,27,28,29). Self-aggregating macromolecules and cytokines are bound to matrix, creating a dynamic microenvironment integral to maintaining tissue homeostasis, which is defined as a state of equilibrium between different yet interdependent elements and which reflects a tissue’s capacity for self-regulation and determination of its cellular composition and structure
Cellular Composition
Fibroblasts and macrophages are present in the interstitial space (23). Cardiac fibroblasts contain messenger RNAs responsible for type I and III collagen gene expression and normal collagen synthesis (30,31). Fibroblasts are also responsible for the synthesis of collagenase and gelatinases, proteolytic enzymes present in the myocardium in latent form (25,32). When activated, these matrix metalloproteinases serve to degrade collagen and to maintain steady-state collagen concentration relative to collagen synthesis. Pluripotent fibroblast-like cells with considerable phenotypic and functional diversity appear under pathological conditions and are responsible for collagen turnover (vide infra).
Cardiac Interstitium in the Infarcted Heart
Fibrosis
Macroscopic scarring of the infarcted myocardium is an expected finding in ICM. Remote to infarct scar(s), microscopic findings include a diffuse perivascular fibrosis of intramural arteries and arterioles; an interstitial fibrosis; foci of microscopic scarring; and a diffuse atrophy of cardiac myocytes (33). In the explanted failing human heart of ischemic origins, multiple foci of microscopic scarring, in combination with interstitial fibrosis, are found in both right and left ventricular myocardium (16). Such remodeling accounts for over two-thirds of all fibrous tissue present, while the infarct scar(s) comprises remaining fibrous tissue
Pathological remodeling of the interstitium in ICM begins with myocyte loss following MI. Its evolution is controlled by events occurring in noninfarcted myocardium involving infarcted and noninfarcted ventricles. This accumulation of extracellular matrix draws attention to tissue repair following myocardial MI.
Fibrogenesis
Repair, a property common to all vascularized tissues, is initiated by inflammatory cells that invade the site of tissue injury. Activated macrophages generate peptides integral to the initiation of repair. Fibroblasts are attracted to the site of infarction and aggregate with macrophages that border on the site of necrotic myocytes and subsequently convert to myofibroblasts (myoFb), as discussed later. Through a complex series of molecular events that include expression of immediate early-response genes and activation of multiple second-messenger systems (which act synergistically to induce mitosis), myoFb proliferate and lay down fibrillar collagen that replaces lost myocytes (34,35,36,37). Cells involved in repair are bathed by tissue fluid whose composition regulates their phenotype and behavior. A diverse array of soluble regulatory signals, whose biological properties are expressed via receptor-ligand binding, gain access to tissue fluid from necrotic myocytes, leukocytes, macrophages, and myoFb. Provided the degree of parenchymal damage is minor, regulatory signals are confined to the site of necrosis. A large, transmural MI, on the other hand, yields a large signal at the site of injury that is widely dispersed within tissue fluid of the common interstitial space of both ventricles to elicit a fibrogenic response in the noninfarcted portion of infarcted and noninfarcted ventricles (17,38,39,40). Signals involved in repair following MI are generally confined to the heart. When they are not, a systemic response (e.g., fever, leukocytosis) appears.
Collagen Turnover
Collagen turnover has been studied in the rat heart both at the site of MI and at remote sites following ligation of the left coronary artery (41,42). At the infarct site and during the early phase of repair, collagen degradation exceeds its synthesis. Matrix metalloproteinases (MMPs) reside in the myocardium in latent form. When activated, MMP-1 (or interstitial collagenase) degrades fibrillar collagen into one quarter and three-quarter fragments; gelatinases (MMP-2 and MMP-9) degrade these smaller fragments. An increase in collagenase activity (Fig. 9-1, left panel) appears at the infarct site on day 2, peaks by day 7, and declines thereafter, together with increased gelatinase activity (not shown) (41). An increase in collagenase (MMP-1) mRNA expression in the infarcted ventricle does not appear until later during week 1, when it replaces the latent pool that is being consumed. Tissue inhibitors of matrix metalloproteins (TIMP) neutralize collagenolytic activity. Transcription of TIMP mRNA at the infarct site peaks on day 2 and declines slowly over the course of 14 days (Fig. 9-1, right panel). Fibroblast-like cells, not inflammatory or endothelial cells, are responsible for the transcription of MMP-1 and TIMP
mRNAs (41). Such events pertaining to collagen degradation are not seen remote to the MI.
mRNAs (41). Such events pertaining to collagen degradation are not seen remote to the MI.
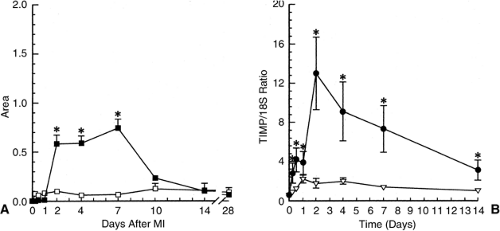 Figure 9-1 A. Scanned lytic areas of the infarcted (solid symbols) and sham-operated rat left ventricle (open symbols). Zymographic data presented for matrix metalloproteinase 1 on days following myocardial infarction. Values are means ±SD; *p <0.05 versus sham-operated animals. B. TIMP and 18S mRNA hybridization after Northern blotting for infarcted (solid symbols) and sham-operated animals (open symbols) rat left ventricle. Values are expressed as the TIMP/18S ratio as means ±SD; *p <0.05 versus sham-operated animals. (Adapted from Cleutjens JPM, Kandala JC, et al. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995;27:1281–1292 , with permission.) |
A fibrogenic component of healing, including an initial expression of fibronectin mRNA (43), accompanies early collagen degradation. Type III procollagen mRNA at the infarct site is increased on day 2 post-MI, reaches a peak by day 21, and declines thereafter (42). Type I procollagen mRNA is increased on day 4 at the site of infarction and remains elevated at week 4 (Fig. 9-2, left panel) and even as late as day 90 (42), suggesting collagen synthesis is an ongoing process and is in keeping with a persistence of an active myoFb phenotype at this site (44,45). A rise in procollagen mRNA is also seen remote to the infarct in the rat model with large, transmural anterior MI (42,46). Procollagen I and III mRNA are increased in the right ventricle and interventricular septum on day 4 and 7, respectively (42). In the septum, closest to the infarct, type I pro-collagen mRNA remains elevated until day 28 (Fig. 9-2, middle panel) while in the right ventricle only until day 7 (Fig. 9-2, right panel). These responses, involving myoFb at the infarct and fibroblasts at remote sites (vide infra) (42,45), are associated with increased expression of transforming growth factor-β1 (TGF-β1) mRNA and its receptors (vide infra) (46).
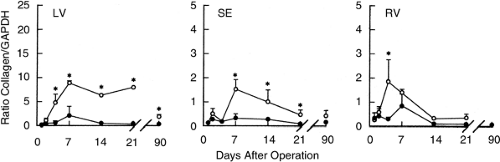 Figure 9-2 Densitometric scanning of type I procollagen versus GAPDH of the autoradiograms after Northern blotting of infarcted left ventricle (LV) and noninfarcted interventricular septum (SE) and right ventricle (RV). Infarcted (open symbols) and sham-operated animals (solid symbols). Values are means ±SD; *p <0.05. (Adapted from Cleutjens JPM, Verluyten MJA, Smits JFM, et al. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338 , with permission.) |
Collagen fibers are first evident at the infarct site on day 7, while an organized assembly of fibers in the form of scar tissue is present by day 14 and continues to accumulate for many weeks (40,47). Hydroxyproline concentration at the site of scarring increases progressively from week 1 to 6, as does collagen cross-linking (48,49,50). A thinning of infarct scar is evident by week 8. Remote to the infarct, including viable left ventricle, interventricular septum, and right
ventricle, fibrillar collagen appears by day 14 and this interstitial fibrosis continues to accumulate for weeks (16,17,38,39,50,51).
ventricle, fibrillar collagen appears by day 14 and this interstitial fibrosis continues to accumulate for weeks (16,17,38,39,50,51).
Cellular Aspects of Interstitial Remodeling
Inflammatory Cells
Repair begins with the formation of a fibrin-fibronectin meshwork and an invasion of injured tissue by inflammatory cells, such as neutrophils and CD4+ lymphocytes that secrete γ-interferon. This serves to activate macrophages (MP) that have invaded the meshwork from the circulation and local sites. Once activated, MP express the gene encoding for inducible nitric oxide synthase and produce nitric oxide, which governs their survival via apoptosis (52,53,54). Activated MP likewise express inducible cyclo-oxygenase that leads to their production of prostaglandins. MP have additional functional activity that includes their ability to generate AngII de novo (55,56). In an autocrine manner, AngII stimulates expression of TGF-β1 that favors the recruitment of pluripotent interstitial fibroblasts and their likely conversion to the myoFb phenotype on day 4 following injury; myoFb proliferation follows. TGF-β1 also serves to suppress the inflammatory cell response. An ACE inhibitor (ACEI) or AT1 receptor antagonist (AT1 Ra), introduced at the time of repair or within the first 48 hours, interferes with the ultimate appearance of fibrous tissue by abrogating this first phase of AngII generation and its important functions in cell-cell signaling and TGF-β1 formation.
Myofibroblasts
MyoFb are central to the fibrogenic component of repair. They appear at sites of injury within days of cardiac myocyte necrosis (40,57,58,59) and are responsible for increased expression of genes encoding for fibrillar type I and III procollagens and their synthesis (40,60,61). Progenitor cells remain to be fully elucidated. Interstitial fibroblasts are likely candidates, not vascular smooth muscle cells or cardiac myocytes (62,63). Fibroblasts have an extensive clonal heterogeneity. MyoFb, phenotypically transformed fibroblastlike cells, have marked functional diversity (62,64) that includes their synthesis of structural proteins and expression of receptors for AngII and TGF-β1. MyoFb also express α-SMA, and these microfilaments endow these cells with contractile behavior. MyoFb are normal residents of heart valve leaflets (45,57,58,65), and leaflet contraction, induced by AngII, is shown in Figure 9-3. MyoFb contraction governs matrix remodeling, including scar thinning (66). The contractile behavior of fibrous tissue is related not only to myoFb having α-SMA, but to their cell-cell and cell-matrix connections. Scar tissue contraction in response to AngII has been demonstrated (67). The contractile nature of fibrous tissue can therefore influence diastolic properties of the infarcted ventricle. The importance of fibrous tissue contraction to ventricular dysfunction seen in ICM remains to be systematically examined.
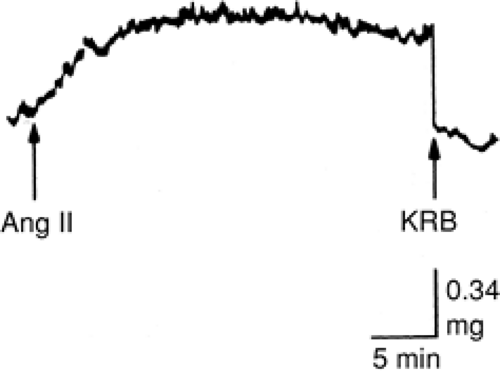 Figure 9-3 Mechanical activity of the anterior rat tricuspid valve leaflet stimulated by 10-5M angiotensin II (AngII). Initial baseline was regained after return to Krebs-Ringer bicarbonate buffer (KRB). Contraction reaches a plateau 3 minutes after stimulation and is maintained for ∼20 minutes. (Adapted from Filip DA, Radu A, Simionescu M. Interstitial cells of the heart valves possess characteristics similar to smooth muscle cells. Circ Res. 1986;59:310–320 , with permission.) |
Signals that determine the appearance of the myoFb phenotype are not entirely certain. TGF-β1 is contributory. Administration of TGF-β1 via a chamber implanted subcutaneously in rats leads to the appearance of myoFb within granulation tissue that forms around the chamber. This was not the case when the chamber contained plateletderived growth factor or tumor necrosis factor-α (68). Cultured adult rat skin fibroblasts undergo a phenotype switch and express α-SMA when incubated with TGF-β1 (68). The appearance of TGF-β1 at sites of MI is likely related to necrotic myocytes (69,70), activated macrophages (71), and myoFb themselves (72).
The fibrillar fibrin-fibronectin scaffolding that forms soon after tissue injury is the precursor to granulation tissue formation and the attachment of myoFb via a fibronexus (73). MyoFb subsequently elaborate type III and then type I collagens, the major fibrillar collagens that constitute fibrous tissue (74,75,76). At pathological sites of tissue repair in the heart, including the site of MI, myoFb express type I and III collagen transcripts (40,42). They elaborate and metabolize substances that regulate their turnover of collagen and govern fibrous tissue contraction in an autocrine manner (vide infra).
A subpopulation of these myoFb are reduced in number at the infarct site via apoptosis (40,57,77). Many, however, persist long after scar tissue formation (44). This contrasts to injured skin, where they completely disappear following repair (78,79). Persistent myoFb in the kidney with experimental glomerulonephritis is associated with a progressive interstitial fibrosis (72). Persistent, active myoFb could contribute to ongoing fibrosis in ICM; this remains to be determined. Irrespective of the location or nature of the inciting stimulus to connective tissue formation in the heart, myoFb are the dominant cell involved in matrix formation at sites of injury (45).
Mediators of Interstitial Remodeling
Transforming Growth Factor-β1
The heart’s healing paradigm is still under investigation. However, it likely does not differ from other organs.
Regulatory peptides, or cytokines such as TGF-β1, contribute to fibrogenesis. TGF-β1 has numerous actions on the extracellular matrix (80), which include fibroblast chemotaxis, phenotype transformation and replication, and ultimately scar tissue formation (68,81). TGF-β1 mRNA is expressed in infarcted myocardium soon after coronary artery ligation (69,70) or catecholamine-induced necrosis (82). Low-density ACE, AngII and TGF-β1 receptor binding, and mRNA expression for type I collagen and TGF-β1 are found in the normal heart. At sites of MI and fibrosis remote to it (including endocardial fibrosis of intraventricular septum), interstitial fibrosis of noninfarcted myocardium and fibrosis of visceral pericardium markedly increased binding of ACE, AngII and TGF-β1 receptors is seen and colocalized with increased type I collagen and TGF-β1 mRNA expression (83). TGF-β1 concentration is increased at each of these sites. TGF-β1 and type I collagen gene expression at each of these sites are attenuated in rats receiving losartan and suggest that locally generated AngII via AT1 receptor binding governs TGF-β1 expression and synthesis at the site of MI and at remote sites in the rat heart.
Regulatory peptides, or cytokines such as TGF-β1, contribute to fibrogenesis. TGF-β1 has numerous actions on the extracellular matrix (80), which include fibroblast chemotaxis, phenotype transformation and replication, and ultimately scar tissue formation (68,81). TGF-β1 mRNA is expressed in infarcted myocardium soon after coronary artery ligation (69,70) or catecholamine-induced necrosis (82). Low-density ACE, AngII and TGF-β1 receptor binding, and mRNA expression for type I collagen and TGF-β1 are found in the normal heart. At sites of MI and fibrosis remote to it (including endocardial fibrosis of intraventricular septum), interstitial fibrosis of noninfarcted myocardium and fibrosis of visceral pericardium markedly increased binding of ACE, AngII and TGF-β1 receptors is seen and colocalized with increased type I collagen and TGF-β1 mRNA expression (83). TGF-β1 concentration is increased at each of these sites. TGF-β1 and type I collagen gene expression at each of these sites are attenuated in rats receiving losartan and suggest that locally generated AngII via AT1 receptor binding governs TGF-β1 expression and synthesis at the site of MI and at remote sites in the rat heart.
TGF-β1 increases transcription of both type I collagen and TIMP by cultured human cardiac fibroblasts (84). AngII augments TGF-β1 gene expression via AT1 receptor binding in cultured neonatal or adult rat cardiac fibroblasts and myoFb (85,86), while endogenous elevations in circulating AngII are associated with upregulation of TGF-β1 gene in the adult rat heart (87). Macrophages, clustered at sites of tissue injury (88), are a likely source of TGF-β1 (71) and, therefore, are important to the appearance of the myoFb (68) and suppression of further inflammatory cell responses (89). A subsequent elaboration of TGF-β1 by myoFb is integral to fibrogenesis, while persistence of myoFb elaborating TGF-β1 leads to progressive fibrosis (72). AngII appears to play a central role in the expression of TGF-β1 by activated macrophages and myoFb. An AT1 Ra prevents these responses (90).
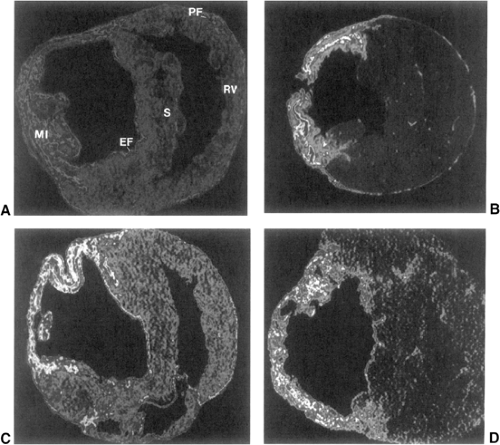 Figure 9-4 Expression of ACE, AngII receptors, type I collagen mRNA, and TGF-β1 mRNA in the infarcted rat heart 4 weeks following coronary artery ligation. By in vitro autoradiography, high-density receptor binding of ACE (A) and AngII (B) is seen at sites of fibrosis including myocardial infarction (MI), endocardium (EF), and pericardium (PF). By in situ hybridization, highdensity binding of type I collagen mRNA (C) and TGF-β1 mRNA (D) is observed at these sites of fibrosis and are colocalized with high-density ACE and AngII receptor binding. Also involved are the interventricular septum (S) and right ventricle (RV). |
Local Angiotensin II
The role of AngII in the fibrogenic phase of tissue repair has been examined in various organs following diverse forms of injury (18). The involvement of this peptide in tissue repair includes its local production at functionally relevant concentrations and the presence of AngII receptors on myoFb responsible for collagen turnover.
Components requisite to the generation of AngII in infarcted rat hearts have been examined. Localization and density of ACE binding in the normal and injured heart (Fig. 9-4) have been addressed using quantitative in vitro autoradiography and an iodinated derivative of lisinopril (125I-351A) (40,45,91,92,93,94,95). In the normal heart, lowdensity ACE binding is found throughout ventricular myocardium and atria. High-density binding is present at sites of high collagen turnover, including heart valve leaflets and the adventitia of intramyocardial coronary arteries and great vessels (95,96,97,98). High-density ACE binding is likewise found in subcutaneous skin with its metabolically active fibroblasts, but not in skeletal muscle tendon with its quiescent fibroblasts (95). Immunolabeling with a monoclonal
ACE antibody (99) identified cells expressing this membrane-bound ectoenzyme: endothelial cells, located on the surface of each valve leaflet; myoFb-like cells residing within leaflet matrix where they are responsible for collagen turnover; and fibroblasts of the adventia of intramural coronary arteries and great vessels. Autoradiography and immunolabeling demonstrated ACE binding in cultured intact myoFb-like cells and their cell membranes (100), where reverse transcriptase-polymerase chain reaction (RT-PCR) demonstrated the presence of ACE mRNA. Substrate utilization of membrane-bound ACE in these cells includes Angl and, in its dual capacity as a kininase II, other chemical mediators of inflammation such as BK and substance P. Thus, myoFb-like cells of valve leaflets have the potential to regulate local concentrations of AngII and other mediators of tissue repair, such as TGF-β1, in valve leaflets. The importance of the TGF-β family of peptides in fetal heart development, including endocardial cushion formation and heart valve induction, has been identified (101,102). A role for AngII generated by these cells in contributing to this process needs to be considered.
ACE antibody (99) identified cells expressing this membrane-bound ectoenzyme: endothelial cells, located on the surface of each valve leaflet; myoFb-like cells residing within leaflet matrix where they are responsible for collagen turnover; and fibroblasts of the adventia of intramural coronary arteries and great vessels. Autoradiography and immunolabeling demonstrated ACE binding in cultured intact myoFb-like cells and their cell membranes (100), where reverse transcriptase-polymerase chain reaction (RT-PCR) demonstrated the presence of ACE mRNA. Substrate utilization of membrane-bound ACE in these cells includes Angl and, in its dual capacity as a kininase II, other chemical mediators of inflammation such as BK and substance P. Thus, myoFb-like cells of valve leaflets have the potential to regulate local concentrations of AngII and other mediators of tissue repair, such as TGF-β1, in valve leaflets. The importance of the TGF-β family of peptides in fetal heart development, including endocardial cushion formation and heart valve induction, has been identified (101,102). A role for AngII generated by these cells in contributing to this process needs to be considered.
High-affinity receptors are integral to the biological activity of ACE-related peptides. The presence of high-density AngII receptor binding in heart valves was demonstrated by autoradiography using 125[Sar1, Ile8] AngII (95). Competitive binding with either an AT1, or AT2 receptor antagonist, losartan and PD123177, respectively, provided identification of AT1 receptors as the dominant subtype. Low-density AT1 receptor binding is present throughout the rat myocardium, while high-density binding is present in heart valve leaflets (95). Western immunoblotting, as well as binding assay, confirmed these findings in myoFb cell membranes (100). Heart valves and myoFb cell membranes like-wise contain BK receptors, as seen by 125I[Tyr8]BK autoradiographic binding and binding assay (95,100).
Cellular responses to AngII receptor-ligand binding has been examined in serum-deprived, cultured myoFb-like cells obtained from adult rat heart valve leaflets. By in situhybridization, these cells express the transcript for type I collagen, and incubation of cultured cells with AngII in pathophysiological concentrations enhances type I collagen synthesis via AT1 receptor binding (100,103,104). Immunohistochemistry (100,105) and electron microscopy (65,106) have shown that these cells contain α-SMA microfilaments. Like vascular smooth muscle cells these microfilaments confer contractile behavior to myoFb in response to various substances that include AngII (65,106).
A biological role for ACE in the local regulation of AngII at normal tissue sites, where collagen turnover is high, is therefore suggested. These findings led to additional autoradiographic and morphological studies in the rat using several different models of experimental injury involving the heart and related structures or systemic organs.
High-density autoradiographic ACE binding was found at the site of MI at week 1 and increased progressively over the course of 8 weeks (Fig. 9-4), in parallel with morphological evidence of fibrillar collagen accumulation found in serial heart sections (40). ACE activity, as measured by substrate conversion, is increased in aneurysmal tissue of the infarcted left ventricle (LV) as contrasted to atrial tissue (107). Additionally, a significant correlation was found between ACE activity and the extent of MI (107), as is also the case for ACE mRNA expression and activity at sites remote to the MI (108). The rat is pronate, which may sustain renal perfusion more effectively than man when in an upright posture. Accordingly, the circulating renin-angiotensin-aldosterone system (RAAS) is not activated in the rat despite extensive MI (108,109,110,111,112).
Monoclonal or polyclonal ACE antibodies were used to identify ACE-labeled cells at sites of healing (40,42,113). Following cardiac myocyte necrosis, these cells include macrophages, α-SMA-positive myoFb, and endothelial cells of the neovasculature. High-density ACE binding is also found at sites of fibrosis remote to MI (Fig. 9-4). This includes endocardial fibrosis of the interventricular septum; interstitial fibrosis of the right ventricle; the fibrosed pericardium that follows pericardiotomy (with or without MI); and the foreign-body fibrosis that surrounds the silk ligature placed around the left coronary artery as part of a sham operation. The anatomic coincidence between high-density ACE binding and fibrosis has been observed in other injured organs of the rat, including the infarcted kidney and incised skin sutured with silk ligature (93). Immunolabeling demonstrated myoFb-containing α-SMA as cells expressing ACE at each of these noninfarcted-related sites. High-density ACE binding is also observed at sites of isoproterenol-induced myocyte necrosis (114,115). At each of these sites of repair, in situ hybridization and immunolabeling demonstrated myoFb as expressing genes encoding for type I and III collagens (40,42).
In vitro quantitative autoradiography was used to address AngII receptor binding in the infarcted rat heart with receptor subtype determined by displacement with an AT1 (losartan) or AT2 (PD 123177) receptor antagonist (105,116,117,118). Marked AT1 receptor binding density is present at the site of MI, as well as endocardial and pericardial fibrosis (Fig. 9-4), while AT2 receptor binding is low at each of these sites. Cells expressing AT1 receptors at these sites of injury are α-SMA-positive myoFb (119). These autoradiographic findings are consistent with the increase in gene transcription and protein expression of the AT1 receptor found in homogenized tissue taken from the site of MI and remote sites using RT-PCR and binding assays (120,121). AT1 Ra abrogates the fibrotic response, which is not the case for AT2 Ra. Increased expression of AngII receptors likewise appears at sites of injury in other organs such as skin (122,123,124).
Using an isolated, crystalloid perfused organ preparation, the conversion of AngI to AngII by epicardium of normal rat hearts was compared to hearts with fibrosis of the visceral pericardium following pericardiotomy alone (without MI) (125). ACE activity of the fibrosed pericardium was several-fold higher than the normal heart and this generation of AngII was completely abrogated by ACEI. High-density ACE binding is likewise present in the perivascular fibrosis and microscopic scars that appear in the right and left atria and ventricles with the chronic administration of AngII or aldosterone (ALDO) (in uninephrectomized rats on a high-sodium diet) (91,126). These latter models, addressed later, demonstrate that ACE binding density within fibrous tissue is independent of circulating AngII or ALDO, in contrast to endothelial ACE of the pulmonary artery, which is subject to negative feedback regulation by circulating AngII (127).
The anatomic coincidence between the expression of ACE and AT1 receptors with normal and pathological expressions of active collagen formation is apparent. High-density ACE and AngII receptor binding are markers of high collagen turnover, as defined by quantitative in situhybridization as high-density, type I collagen expression. Alpha-SMA-containing, fibroblast-like cells, normally found within valve leaflets and vascular adventitia, and pathological sites of fibrous tissue formation express genes encoding for ACE, AT1 receptors, and fibrillar collagens. MyoFb could therefore be considered a “metabolic entity” regulating their own collagen turnover (128).
ACE binding density correlates with the presence of myoFb. The disappearance of ACE-positive cells or a reduction in their absolute number reduces ACE binding density at sites of fibrosis, as is the case with some sarcoid granulomas (129). Both ACE and Angll receptor binding densities in the infarcted rat heart remain high for months post-MI (40,116), as does ACE activity (108). Each is in keeping with the persistence of myoFb at the infarct site (44).
Granulation tissue and its diverse cell populations, in particular, generate peptides integral to tissue repair via receptor-ligand binding. ACE, for example, regulates local concentrations of AngII. At the infarct site the concentration of AngII is increased several-fold above that found in viable myocardium (110). De novo Ang peptide generation requires expression of several requisite components: angiotensinogen (Ao), the precursor to all Ang peptides; a protease that cleaves amino acids to form AngI; and ACE that hydrolyzes the decapeptide AngI to AngII, an octapeptide. An independent pathway of AngII generation involving a chymase has been suggested; however, increased expression of its transcript is infrequently found in the failing human heart (130).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


