Fig. 2.1
Regulation of mitophagy and autophagy. Schematic summarizes the p62/SQSTM1-regulated formation of autophagosome for recycling the damaged mitochondria and misfolded and aggregated proteins. Autophagy and mitophagy proceed through several phages including initiation, elongation, maturation, cargo sequestration, and autophagosome and lysosome fusion. p62 recruits LC3 to the autophagosome membranes that is required for the autophagosome maturation
Mitophagy targets depolarized mitochondria, notably during apoptosis. It results in the formation of an isolation membrane enclosing organelles and fragments of the cytoplasm that are delivered into lysosomes for hydrolytic digestion and recycling. Mitophagy ensures that damaged or excessive mitochondria are properly directed toward degradation via autophagy [47]. Thus, autophagy is important for recycling intracellular components for cell survival and also essential for maintaining organelle homeostasis and quality. Not only is autophagy important for mitochondrial function, but also there is growing body of evidence that the mitochondrion itself plays a significant role in autophagy. For example, mitochondrial dysfunction has been associated with neurodegenerative disease such as Parkinson’s and also with some metabolic disorders [51]. The mitochondria and the process of autophagy are uniquely related, in that defects in either can increase the risk of certain metabolic and autophagic diseases [52]. Further investigations of the functional interactions between mitochondrial and autophagy functions and dysfunctions would reveal the mechanisms underlying pathogenesis of various diseases.
Mitochondrial Dysfunction in Lung Diseases
Mitochondria act as sensors of oxidative stress and a focal point of cellular signaling platforms, especially those involved in modulating cell death, including necrosis, apoptosis, and autophagy [53–57]. A deficiency in energy metabolism, the bioenergetic failure characteristic of mitochondrial disease states [58], has been implicated in a variety of human diseases. Various diseases, such as glaucoma, inflammation, neurodegenerative diseases, type 2 diabetes, cardiomyopathies, dysrhythmias, and cancers, especially those involving prostate and colon, have been linked to mitochondrial dysfunction (reviewed in [59]). All these diseases have been associated with defects in mitochondrial function [60–63] or with an inability to accommodate the consequences of oxidative stress [64]. Although ROS act as signaling molecules at low/physiological levels, excessive ROS production causes nuclear DNA damage and the degradation of oxidized proteins, lipids, and nucleic acids. It also inflicts damage on various cellular organelles, particularly mitochondria [65]. The spatial proximity of mtDNA to the free radicals produced by the electron transport chain (ETC) makes it uniquely susceptible to mutations, especially when the ETC becomes dysfunctional [66]. The current view is that ROS are a primary cause of mitochondrion-driven diseases and mitochondrial dysfunction, and the resulting oxidative damage can have profound effects, such as a buildup of cytotoxic metabolites, energy depletion, and even cell death and organ dysfunction [12, 67].
During development, in high-altitude travel, and in acute and chronic lung disease states, lung cells are exposed to hypoxia, and the release of ROS from the inner mitochondrial membrane is triggered by hypoxic conditions. The ETC serves as the critical cellular oxygen sensor for many of these responses. Although ROS signals generated by hypoxia are thought to play a crucial role in pulmonary development in the newborn and produce responses in mature lungs (primarily guarding cells from hypoxic injury), unremitting ROS production and signaling are counterproductive, because they inflict cellular damage and degrade the normal function of the lungs [68]. The lungs of newborns and adults are constantly exposed to various environmental stressors that cause epithelial and endothelial cell dysfunction and death, leading to acute lung injury and inflammation and respiratory impairment. A prominent role has been suggested for mitochondrial dysfunction in acute and chronic lung disorders. For example, damage to mitochondria was observed in the lungs of rats subjected to ischemic reperfusion, and this damage was accompanied by tissue injury and inflammation [69]. Blocking mitochondrial complex III activity reduces LPS-induced, ROS-mediated acute lung injury, pointing to a central role for mitochondrial ROS in promoting pulmonary disorders [70]. Exposure of mice to LPS causes oxidative mitochondrial damage and biogenesis in cardiomyocytes, and LPS-induced lung injury is accompanied by mitochondrial biogenesis, suggesting that this process is important for tissue repair and resolution [71]. Decreased levels of mitochondrial glucocorticoid (mtGR), estrogen (mtER) receptors, and mitochondrial oxidative phosphorylation (OXPHOS) enzyme biosynthesis have been observed in the lungs of mice with allergic airway inflammation, suggesting that either a loss or decreased function of these proteins contributes to airway diseases such as asthma [72]. Genetic mutations in SARS2, which encodes mitochondrial seryl-tRNA synthetase, have been found in infants with hyperuricemia, pulmonary hypertension, renal failure, and alkalosis (HUPRA) syndrome and are accompanied by a lack of acylated tRNA [73]. Prohibitins (PHB1 and PHB2) form large, multimeric ring complexes in the inner membrane of mitochondria and interact with the NADH dehydrogenase protein complex, constituting an essential pathway for the mitochondria [74]. Knockdown of PHB1 or PHB2 leads to mitochondrial damage and dysfunction, accompanied by enhanced levels of ROS generation in adipocytes [75]. Prohibitin expression was shown to be downregulated in the lungs of COPD and non-COPD smokers, suggestive of an alteration of mitochondrial function, possibly because of decreased mitochondrial stability caused by cigarette smoke exposure. This downregulation may be causally linked to genesis of COPD [76]. Further studies are warranted to better define the exact mechanisms of regulation and the functions of nuclear and mitochondrial genes encoding proteins that control mitochondrial functions in the lungs during development and to characterize the physiological conditions and pathological states caused by environmental stressors.
Several studies have explored the use of small molecules, peptides, and proteins that target mitochondrial ROS as part of a strategy to mitigate cellular stress and tissue damage, and they have displayed beneficial effects in culture systems and animal models of lung diseases. For example, oxidant stress causes mtDNA damage and death in pulmonary artery endothelial cells, but overexpression of a DNA repair enzyme, Ogg1, mitigates mtDNA damage and cytotoxicity [77]. Pretreatment of rodents with antioxidants suppresses oxidant-induced lung pathogenesis, but their use to improve the outcomes of patients in the clinical setting has had only limited beneficial effects, if any [78–80]. Nevertheless, new and novel approaches are being evaluated to improve human health, with specific emphasis on enhancing mitochondrial functions. For example, a new study using an LPS-induced model of acute lung injury has demonstrated that bone marrow-derived stromal cells can repair tissue injury through the transfer of mitochondria [81]. This mitochondrial transfer resulted in increased levels of alveolar ATP concentrations, suggesting that mitochondrial transfer can rescue and repair injured cells, a finding that could have important implications for a variety of diseases linked to abnormal tissue repair [81].
Regulation of Mitochondrial Metabolism and Functions by the NRF2-ARE Pathway
NRF2 is a cap‘n’collar basic leucine zipper transcription factor. The Kelch-like ECH-associated protein 1 (KEAP1) retains NRF2 in the cytoplasm and promotes its proteasomal degradation [82]. Several stressful and electrophilic stimuli are known to disrupt KEAP1/NRF2 interactions, leading to the release of NRF2 from the cytoplasm and its subsequent nuclear accumulation [83–85]. In the nucleus, NRF2 heterodimerizes mainly with the MAF (Maf-G, Maf-F, and Maf-K), JUN (c-Jun, Jun-B, and Jun-D), and ATF (ATF-4) families of bZIP proteins prior to binding to the DNA sequence 5′-TGAG/CnnnGC-3′ (the antioxidant response element (ARE)), resulting in the transactivation of a network of genes that encode cytoprotective and antioxidative enzymes and proteins [83, 86]. Some of the well-characterized targets include GPX2, NQO1, GCLC, and GCLM, HMOX1. Further, NRF2 also regulates the expression of nuclear genes that encode several mitochondrial proteins (see Table 2.1) [87–100]. As described above, nuclear respiratory transcription factor NRF-1 regulates mitochondrial gene expression by inducing TFAM.
Table 2.1
NRF2-regulated mitochondrial proteins encoded by nuclear DNA
Gene name | Gene symbol | References |
|---|---|---|
Aldo-keto reductase | AKR1B | Nishinaka et al. [87] |
Cystine-glutamate transporter | xCT | Sasaki et al. [88] |
Glutathione S-transferase alpha 3 | GSTα3 | Tjalkens et al. [89] |
Glutathione S-transferase alpha 4 | GSTα4 | Hayes et al. [90] |
Glutathione S-transferase mu1 | GSTmu1 | Tjalkens et al. [89] |
Glutathione S-transferase pi1 | GSTpi1 | Ikeda et al. [91] |
Glyoxalase 1 | GLO1 | Xue et al. [92] |
Malic enzyme 1 | ME1 | Mitsuishi et al. [93] |
Isocitrate dehydrogenase 1 | IDH1 | Mitsuishi et al. [93] |
Nuclear respiratory factor 1 | NRF-1 | Piantadosi et al. [94] |
Peroxiredoxin 1 | PRDX1 | Kim et al. [95] |
Peroxiredoxin 3 | PRDX3 | Miyamoto et al. [96] |
Peroxiredoxin 5 | PRDX5 | Miyamoto et al. [96] |
Sulfiredoxin | SRX | Soriano et al. [97] |
Superoxide dismutase 1 | SOD1 | Dreger et al. [98] |
Thioredoxin | TRX1 | Kin et al. [99] |
Thioredoxin reductase 1 | TXNRD1 | Sakurai et al. [100] |
The promoter of NRF-1 has AREs, the binding sites for NRF2 [94]. Activation of NRF2 enhances mitochondrial biogenesis through transcriptional activation of the NRF-1 promoter [94] (Fig. 2.2). Piantadosi et al. have demonstrated that carbon monoxide (CO), a product of HMOX1 activity, elevates the mtDNA copy number in an NRF2-dependent manner in the lungs of mice subjected to pneumonia [94]. This group has shown that CO elevates mitochondrial H2O2 production, especially which in turn activates the PKB/AKT kinase. AKT then phosphorylates (and thus inactivates) GSK-3β, allowing NRF2 nuclear accumulation and potentiation of NRF2-depenent gene expression [101]. By inducing mitochondrial autophagy, especially when the damage to mitochondria caused by oxidative stress is irreparable, HMOX1 confers protection to lung epithelial cells exposed to chronic levels of hyperoxic stress, perhaps by decreasing the abnormal levels of ROS generated by damaged mitochondria [102]. The NRF2-ARE pathway therefore plays a key role in guarding mitochondria against oxidative stress. Recent evidence shows that mitochondrial ROS activate downstream protective mechanisms including the NRF2-ARE pathway [103, 104]. Mitochondrial phosphoglycerate mutase family member 5 (PGAM5), a protein phosphatase, plays an important role in regulating mitochondrial functions. When targeted to the outer membrane of mitochondria, forms a ternary complex with the KEAP1-NRF2 dimer [105], and knockdown of either KEAP1 or PGAM5 activates NRF2-dependent antioxidant gene expression in HeLa cells, suggesting that PGAM5 regulates mitochondrial functions by modulating NRF2 activity indirectly via KEAP1[105]. KEAP1 via PGAM5 promotes degradation of antiapoptotic proteins BCL-Xl and BCL2 [106], whereas NRF2 upregulates antiapoptotic protein BCL2 [107]. Thus, PGAM5/KEAP1/NRF2 interactions add another dimension to NRF2 regulation and functions during cellular injury and death, because PGAM5 acts as a crucial player in promoting necrotic cell death [108]. It would be important to determine under what conditions the KEAP1-NRF2 complex tethered to mitochondrial membrane by PGAM5, the relevance of PGAM5/KEAP1/NRF2 interactions during stressful insults, and how oxidative stress perturbs these interactions favoring mitochondrial dysfunction and autophagy or cell death in response to chronic stress and in disease states (Fig. 2.3).
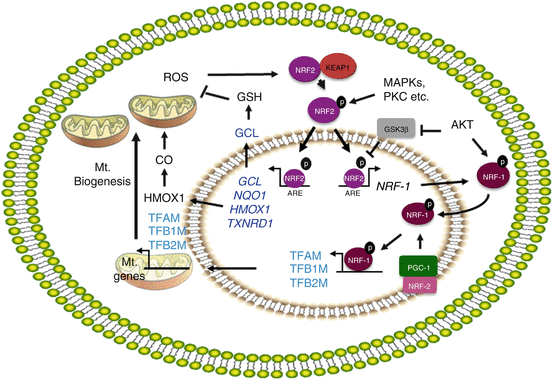
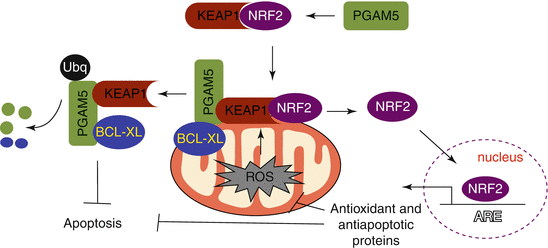
Fig. 2.2
Regulation of mitochondrial biogenesis by N2. Schema summarizes the signaling- and NRF2-regulated proteins involved in countering the ROS-mediated stress and mitochondrial (Mt) biogenesis. HMOX1 generates CO by degrading heme, and CO stimulates ROS generation in mitochondria. ROS oxidize KEAP1 cysteine residues and release NRF2 from KEAP1 and phosphorylated by MAP kinases/protein kinase C. NRF2 then translocates into the nucleus and transcriptionally activates the genes that encode antioxidant proteins and NRF-1. NRF-1 upon activation by AKT translocates into the nucleus and further activated by PGC-1 family members and NRF-2. NRF-1 activates several nuclear-encoded mitochondrial genes including mitochondrial transcriptional factors, TFAM, TFB1M, and TFB2M and promotes mitochondrial biogenesis. The arrows represent activation, and “blunted” arrows represent inhibition
Fig. 2.3
PGAM5-regulated KEAP1-NRF2 interactions and antioxidant response. PGAM5 tethers KEAP1/NRF2 complex to the mitochondrial membrane during mitochondrial stress. Mitochondrial ROS cause dissociation of NRF2 from KEAP1. NRF2 enters into the nucleus and activates antioxidant and antiapoptotic gene expression to mitigate mitochondrial stress. KEAP1 targets PGAM5 and BCL-XL proteins to proteasomal degradation and inhibits apoptosis
NRF2-Regulated GSH Signaling in Mitochondrial Metabolism and Function
NRF2-regulated transcriptional activation of GCLC and GCLM is crucial for de novo synthesis of GSH, the most abundant nonprotein thiol in the cell, in both constitutive and induced states. Oxidative and electrophilic stress induces de novo GSH synthesis via NRF2-mediated upregulation of the xCT anionic amino acid transporter [109]. Thus, NRF2 plays key roles in mitochondrial metabolism and protection by regulating the expression levels of genes that regulate GSH biosynthesis (GCLC, GCLM, and xCT or SCLA711) and turnover (see below). Under normal conditions, GSH is synthesized in the cytosol and then relatively slowly transported into the mitochondria [110, 111]. The mitochondrial GSH (mGSH) pool is maintained at 1–5 mM, even when the cytosolic redox balance is disturbed [112, 113]. This concentration is regulated by the action of glutathione reductase (GR) [114, 115], which reduces mGSSG to mGSH using NADPH. GPx, in the presence of GSH, detoxifies H2O2, leading to the generation of H2O and GSSG. There are two forms of GPx in the matrix: Soluble GPx1 [116] mainly degrades H2O2, whereas GPx4, located on the matrix surface of the inner membrane, degrades phospholipid hydroperoxides and organic hydroperoxides to alcohols [117–119]. GR then converts GSSG to GSH at the expense of NADPH in order to prevent the loss of GSH. The GSH/GSSG redox couple is an example of a major thiol/disulfide couple in the cell that helps to maintain the overall redox state. Studies have found the GSH/GSSG redox potential inside the mitochondria to be slightly more negative (approximately −280 mV) than the cytoplasm, indicating a more reduced environment [120, 121]. However, much remains unclear about the nature and regulation of mGSH transport and the means by which mitochondria maintain GSH in a reduced form.
The reversible S-glutathionylation of mitochondrial enzymes has emerged as an important mechanism for the regulation of metabolism in response to changes in redox environment and ROS production [122]. The mitochondrial matrix environment, with its basic (pH ~8) conditions and high concentration of GSH [122, 123], provides the necessary milieu to promote thiol residue modification of proteins by GSH. This process has been suggested to act as a protective mechanism to prevent protein oxidation/deactivation, and it also serves as a negative regulator of ROS production from complex I [124]. Glutathionylation also plays a protective role after oxidative stress or ischemia preconditioning because it protects exposed cysteines from oxidative damage. During oxidative stress, the thiols of several proteins undergo reversible oxidation to sulfinic (SO2H) or sulfonic acid (SO3H) [125–127]. Hence, by reversibly modifying critical thiols, glutathionylation can protect enzymes in the mitochondria during oxidative stress.
Glutaredoxins (GRXs) catalyze the deglutathionylation of protein-GSH mixed disulfides far more effectively than do other thiol proteins such as thioredoxin (TRX) [128]. Mitochondrial GRX2 has a CSYC motif in its active site, with cysteine 70 playing a key role in deglutathionylation [129–131]. GRX1 has an additional exposed cysteine that is readily modified by oxidants; however, GRX2 lacks this cysteine residue, which may make it less easily inactivated by oxidative stress, S-nitrosating agents, and GSSG within the mitochondrial matrix [129, 131, 132]. Furthermore, GRX2 can be reduced directly by thioredoxin reductase 2 (TXNRD2) [132, 133]; however, this reduction is far less efficient than reduction by GSH, which is more effective even at a GSH level that is 10 % of normal [133]; consequently, the TXNRD2 is unlikely to play a physiologically important role in GRX2 reduction. Together, these factors appear to enable GRX2 to operate more effectively than GRX1 in an environment with more oxidative stress. GRX2 can form an inactive dimer around an iron sulfur center, and this dimer formation can be reversed by oxidative stress, potentially enabling the activation of GRX2 in response to elevated mitochondrial oxidative stress [134–136]. Recently, it has been demonstrated that glutathionylation and deglutathionylation play a key role in controlling ROS-induced proton leakage through uncoupling proteins, with glutathionylation decreasing proton leak-dependent oxygen consumption in mitochondria isolated from skeletal muscle [137]. It would be of great interest to determine the exact relevance and mechanisms of glutathionylation and deglutathionylation balance during mitochondrial functions and dysfunction in acute and chronic lung disease states.
NRF2 Impairment and Mitochondrial Dysfunction
Levels of mitochondrial ROS are counterbalanced by NRF2-regulated antioxidant enzymes and proteins. Some of these enzymes and proteins include HO1 [138], NQO1 [139], GST [140], solute carrier family 7 (SLC7A11) [141], GSH [142], TXNRD1 [100], and PRDX [141]. Recent studies have demonstrated that NRF2 also regulates expression of the genes encoding proteins that control cellular anabolic metabolism [93]. Dysregulation of the expression of these antioxidative and metabolic enzymes and proteins promotes enhanced oxidative stress, mitochondrial damage, and impairment of mitochondrial function, leading to lung disease states. Consistent with this view, several studies (including ours) using experimental models of stress-induced pulmonary disorders that are known to cause mitochondrial dysfunction have shown that NRF2 confers protection against endotoxin-induced lung injury and septic shock [143, 144], pro-oxidant-induced lung injury and fibrosis [145, 146], and cigarette smoke-induced emphysema [147]. Loss of NRF2 causes a redox imbalance primarily as the result of a diminished or low level expression of genes encoding several antioxidative and cytoprotective enzymes in both the constitutive (basal) states and in response to stressful stimuli in lung-resident cells and alveolar inflammatory cells [83, 86, 148, 149]. Loss of NRF2 also impairs the resolution of sublethal hyperoxic lung injury and inflammation, but supplementation of Nrf2-null mice with GSH following lung injury can improve the resolution of their lung damage [150]. Nrf2-deficient pups, when exposed to hyperoxia at birth for four days, develop greater levels of alveolar simplification (septal growth arrest) than do Nrf2-sufficient pups [151]. Nrf2 deficiency enhances cellular stress and susceptibility to oxidant-induced lung epithelial cell death [152], and its overexpression confers cellular protection against hyperoxia in lung epithelial cells [148, 153], as well against pro-apoptotic stimuli in non-lung epithelial cells [85, 154]. This experimental evidence strongly supports an important role for the NRF2-driven transcriptional response in mitigating cellular stress and mitochondrial functions induced by pro-oxidants and toxicants.
In agreement with the experimental studies described above, a decline in NRF2-regulated cytoprotective gene expression has also been observed in the lungs of patients with COPD, and this loss is associated with the decreased expression of DJ-1 [155], which is known to confer protection against oxidative stress by regulating mitochondrial ROS and functions [156]. An association between NRF2 polymorphisms and an increased risk of susceptibility to acute lung injury (ALI) and decline of lung function in smokers has been reported [157–159]. Pharmacological activation of NRF2 improves the antibacterial defenses of alveolar macrophages in patients with COPD [160] and restores corticosteroid responses in patients with COPD [161], suggesting the involvement of a dysfunctional NRF2 or an inactivation of NRF2 signaling in bacterial exacerbations in smokers. The levels of NRF2 signaling and NRF2-dependent genes are altered in patients with mitochondrial diseases [162], suggesting that decreased NRF2 activity is associated with the progression of mitochondrial diseases. Nuclear translocation of NRF2 has been shown to be impaired in neurodegenerative diseases such as Alzheimer’s (AD) and Parkinson’s disease (PD), and dysfunction in the NRF2 pathway has been shown to lead to a decreased cellular defense against oxidative stress [163]. In certain disease states such as diabetes, mitochondrial GSH levels are decreased [112, 113, 164]; GSH depletion decreases mitochondrial antioxidant defenses in diabetic cardiomyocytes and renders them more sensitive to apoptosis after an oxidant insult [112]. These findings may explain why the elderly with diminished NRF2 activity [165] and endogenous antioxidant defenses have a higher incidence of type 2 diabetes and cardiovascular problems. Thus, targeting NRF2 can be a novel therapeutic approach to addressing multifaceted antioxidative and innate responses in order to counteract oxidative stress-induced and mitochondria-mediated lung disorders and other disease states.
Activation of the NRF2 Pathway as a Therapeutic Approach to ameliorate Mitochondrial Dysfunction
Several compounds that specifically disrupt KEAP1/NRF2 interactions, such as isothiocyanates, sulforaphane, indoles, and triterpenoids, confer protection against ALI and inflammation in preclinical models of human disorders, and this protective response has been shown to be correlated with an elevated level of antioxidant gene expression regulated by NRF2 [166, 167]. Notably, triterpenoid analogs are more potent than isothiocyanates and sulforaphane in inducing the NRF2-ARE-mediated transcriptional response, and these compounds have been shown to have beneficial effects at nanomolar concentrations in various preclinical models of tissue injury and inflammation (see review [168]). For example, mice administered with the triterpenoid compound CDDO-Im (1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole) show decreased levels of hyperoxia- [169] and LPS-induced lung injury and inflammation in mice [170], as well as reduced cigarette smoke-induced emphysema [171], when compared to their vehicle-treated counterparts. This protection by CDDO-Im was observed in Nrf2-sufficient but not Nrf2-deficient mice, suggesting that specific targeting of NRF2-ARE signaling may provide a novel therapeutic strategy for treating human diseases. While triterpenoids have shown significant positive outcomes in various preclinical models, a recent clinical study with bardoxolone, an CDDO analog, showed adverse cardiovascular effects in phase III trial of diabetics with severe stage 4 chronic kidney disease, although it improved kidney function [172]. Further refinement of existing NRF2 activators and/or the development of novel activators are clearly needed in order to enhance NRF2-mediated mitochondrial biogenesis and function and improve disease outcomes.
Summary and Future Perspectives
The mitochondria-dependent release of ROS is essential for various physiological responses, such as adaptation to cell growth conditions and variations in oxygen levels. Elevated levels of mitochondrial ROS generated in response to environmental stress, genetic, and metabolic state feedback are known to cause mitochondrial dysfunction, resulting in organ dysfunction and ultimately a pathological state. NRF2-driven ARE-mediated transcriptional responses are crucial for maintaining intracellular redox homeostasis, and an NRF2 deficiency increases susceptibility to the development of lung pathogenesis in experimental animals. Consistent with these experimental data, NRF2/ARE dysfunction has been reported in human lung diseases, including COPD and pulmonary disorders. NRF2 deficiency impairs the resolution of lung inflammation and tissue repair and increases susceptibility to pathogenic infections following exposure to oxidants. Mitochondrial dysfunction has been implicated in all these processes. Recent studies have revealed that NRF2 regulates mitochondrial biogenesis by inducing the gene coding TFAM, a transcription factor that binds to mitochondrial gene promoters and induces their expression, and by upregulating HMOX1 expression. Because oxidative stress caused by mitochondrial dysfunction can lead to NRF2 activation, mitochondria damage, and mitophagy, it is likely that NRF2 activation acts as an autoregulatory feed-forward loop to dampen the increased ROS levels and to promote mitochondrial biogenesis, thereby maintaining homeostasis following tissue or cellular injury. Recent studies have shown that NRF2 promotes autophagy, indicating that NRF2 modulates mitophagy, rather than mitochondrial biogenesis, under conditions of oxidative damage to the mitochondria. It is unclear whether the impairment of NRF2-ARE signaling contributes to the subsequent pathogenesis caused by mitochondrial dysfunction and likewise whether mitochondrial dysfunction compromises NRF2-ARE signaling and leads to pulmonary disease. Compounds that target NRF2-ARE signaling have shown promising results in experimental models of ALI/ARDS, septic shock, and emphysema. The use of new small-molecule activators that specifically target and upregulate the NRF2-ARE transcriptional response may be a better therapeutic option for blocking cellular stress and promoting mitochondrial biogenesis in diseases linked to mitochondrial dysfunction. Indeed, compounds that target NRF2-ARE signaling are in phase III clinic trails to improve clinical outcomes in patients with various diseases, such as COPD. Although the results are promising, whether these compounds provide a significant improvement in clinical outcomes in critically ill patients remains to be seen.
Acknowledgments
Supported by the NIH grants HL66109, ES11863, DK084445, and ES18998 and the Flight Attendant Medical Research Institute (FAMRI) (to SPR) and NIH grant CA78282 (to DVK).
References
1.
Wittig I, Schagger H. Supramolecular organization of ATP synthase and respiratory chain in mitochondrial membranes. Biochim Biophys Acta. 2009;1787:672–80.PubMed
2.
Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396–402.PubMed
3.
Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci. 2000;25:319–24.PubMed
4.
Duchen MR. Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol Aspects Med. 2004;25:365–451.PubMed
5.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13.PubMedCentralPubMed
6.
Murphy MP. Mitochondria–a neglected drug target. Curr Opin Investig Drugs. 2009;10: 1022–4.PubMed
7.
West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–80.PubMedCentralPubMed
8.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5.PubMed
9.
Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn 2nd GW, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. 2011;108:837–46.PubMedCentralPubMed
10.
Leloup C, Tourrel-Cuzin C, Magnan C, Karaca M, Castel J, Carneiro L, Colombani AL, Ktorza A, Casteilla L, Penicaud L. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes. 2009;58:673–81.PubMedCentralPubMed
11.
Bonawitz ND, Chatenay-Lapointe M, Pan Y, Shadel GS. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007;5:265–77.PubMedCentralPubMed
12.
Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441:523–40.PubMedCentralPubMed
13.
Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–93.PubMed
14.
Henze K, Martin W. Evolutionary biology: essence of mitochondria. Nature. 2003;426:127–8.PubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


