Chapter 54 Rare Diffuse Interstitial Lung Diseases
Rare Cystic Lung Diseases
Lymphangioleiomyomatosis
Lymphangioleiomyomatosis (LAM) is a cystic lung disease that affects almost exclusively women and has a prevalence of approximately 5 women per 1 million of most populations. LAM may occur sporadically or as part of the autosomal dominant genetic disorder, tuberous sclerosis complex (TSC). In both forms the lungs and lymphatics are infiltrated by LAM cells, an abnormal cell clone harboring biallelic inactivating mutations in the genes associated with TSC, either TSC-1 or more often TSC-2. LAM cells accumulate and cause progressive cystic destruction of the lungs, probably by the elaboration of proteolytic enzymes (Figure 54-1). LAM cells form small nodular clumps associated with lymphatic endothelial cells, which in turn form lymphatic channels that allow LAM cells to disseminate throughout the body. LAM cells also infiltrate the axial lymphatics and form the smooth muscle elements of angiomyolipoma, a rare tumor of the perivascular epithelioid cell family, present in up to 40% of women with sporadic LAM and almost all patients with TSC-LAM.
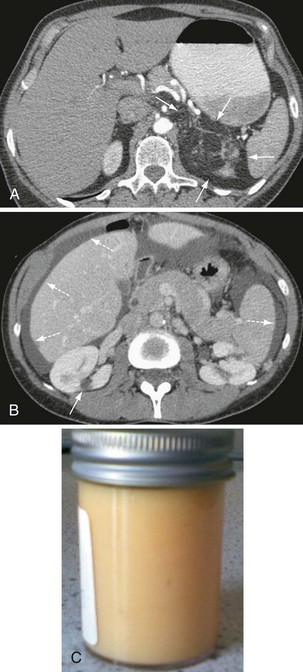
Other, extrapulmonary manifestations are related to lymphatic obstruction and include lymphadenopathy in up to 30% of patients and lymphangioleiomyomas, cystic lymphatic swellings usually in the abdomen, pelvis, or retroperitoneum occurring in up to 20% of patients (Figure 54-2). Thoracic and abdominal chylous collections are present in up to 10% of patients. LAM is also associated with a higher prevalence of meningioma than the general population. Cystic lung destruction results in recurrent pneumothorax and progressive airflow obstruction, with an average decline in forced expiratory volume in 1 second (FEV1) of 120 mL per year (normal, ~27 mL/yr). However, the clinical course of patients with LAM varies, with some remaining stable for many years. At present, no features definitively predict which patients will develop progressive disease; however, onset at a younger age, presentation with breathlessness, and poor lung function at presentation suggest more aggressive disease.
Langerhans cell histiocytosis
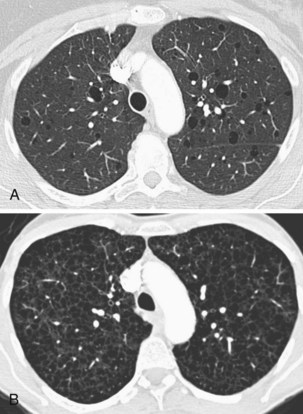
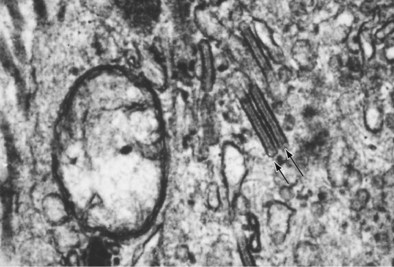
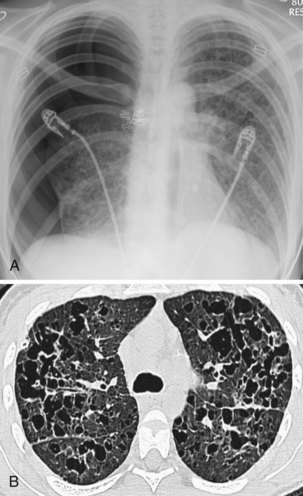
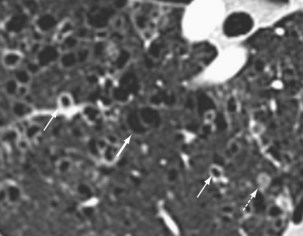
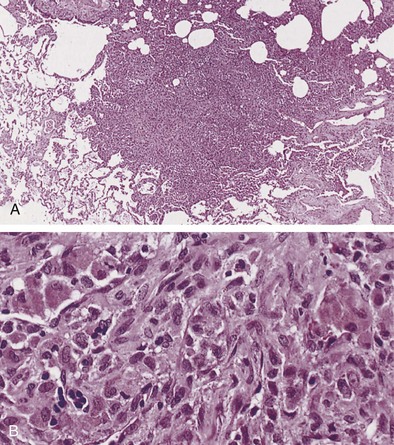
Langerhans cell histiocytosis typically affects individuals between 20 and 40 years of age, almost all of whom are current or recent smokers. The infiltrating Langerhans cell clone expresses the surface protein CD1a and has a characteristic appearance on electron microscopy with Birbeck inclusion granules (Figure 54-3). LCH may coexist with areas of desquamative interstitial pneumonitis, another smoking-related entity characterized by activation of macrophages. Similar to LAM, LCH is of variable severity, with approximately one quarter of LCH patients detected by imaging being asymptomatic. Pneumothorax is the presenting feature in about 20% of patients; the rest present with cough, dyspnea, or weight loss. The chest x-ray film is abnormal in most patients, showing reticular shadowing with midzone and upper-zone predominance (Figure 54-4). The CT scan shows a mixture of nodules, cavitating nodules, and cysts, often with thick, irregular walls forming bizarre shapes (Figure 54-5); characteristically, the bases of the lungs are relatively spared. As the disease progresses, the cysts tend to amalgamate, and nodules become less common. The diagnosis is usually suspected by the combination of these imaging appearances in younger smokers. Absolute confirmation often requires a surgical biopsy (Figure 54-6). However, bronchoalveolar lavage (BAL) fluid with greater than 5% Langerhans cells identified by CD1a positivity, often with increased macrophages and eosinophils, is supportive and can be diagnostic in the correct clinical context.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


