10 Pulmonary Vasculitis and Pulmonary Hemorrhage
Pulmonary vasculitis
Overview of Pulmonary Vasculitis
Inflammation of arteries and veins can occur in many inflammatory lung diseases, including infections. By convention, the diagnostic term pulmonary vasculitis is restricted to a relatively limited number of diseases in which vascular inflammation is thought to be a major component of the pathologic process. Most pulmonary vasculitides are believed to be immune-mediated diseases, although their etiology and pathogenesis remain unknown. By best estimates, the overall annual incidence of the major forms of vasculitis is 39 per 1 million.1
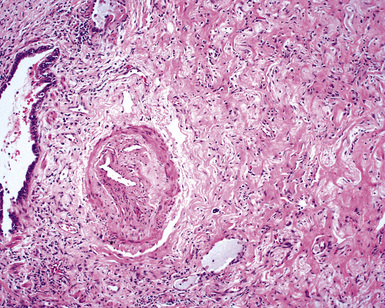
When pulmonary vasculitis occurs, there is inflammation of the vessel wall, often accompanied by fibrin and sometimes, necrosis. Cuffing of blood vessels by inflammatory cells, a nonspecific finding, must be distinguished from infiltration of inflammatory cells into the media and intima of arteries and veins (Fig. 10-1).
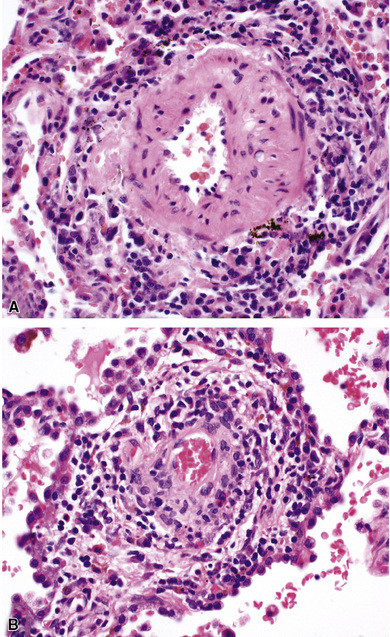
A diagnosis of pulmonary vasculitis carries a strong implication for immediate therapeutic intervention (typically immunosuppression) and therefore should never be made lightly. Furthermore, serologic and clinical correlation with the pathologic findings is essential for a correct diagnosis. Typical histologic examples of pulmonary vasculitis-capillaritis are shown in Figure 10-2.
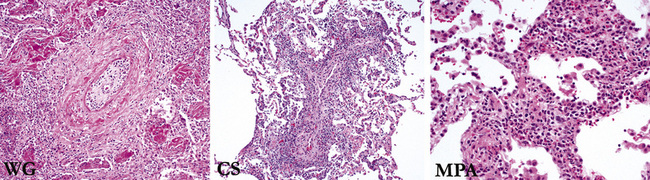
The general category of pulmonary vasculitis includes a number of different diseases that can be more easily understood by dividing them into three main groups: (1) idiopathic vasculitic syndromes that commonly involve the lung (e.g., Wegener granulomatosis [WG]), (2) vasculitic disorders that rarely involve the lung (a much larger number), and (3) miscellaneous conditions that produce pulmonary vascular inflammation2,3 (Box 10-1).
Box 10-1 Pulmonary Vasculitis Syndromes
Modified from Travis W, Koss M. Vasculitis. In: Dail D, Hammar S, eds. Pulmonary Pathology. New York: Springer-Verlag; 1994:1027–1095.
WG, Churg-Strauss syndrome (CSS), and microscopic polyangiitis are the idiopathic vasculitis syndromes that commonly affect the lung. The conditions of bronchocentric granulomatosis and lymphomatoid granulomatosis traditionally have been grouped in the category of pulmonary “angiitis and granulomatosis”; however, neither of these entities is currently thought to be a vasculitic condition. Bronchocentric granulomatosis is a morphologic pattern of airway inflammation that occurs in a variety of conditions, especially infection, and lymphomatoid granulomatosis (also known as angiocentric immunoproliferative disorder) is now known to represent a lymphoproliferative disease in which prominent vascular involvement occurs.4–6
Idiopathic Vasculitic Syndromes That Commonly Affect the Lung
Wegener Granulomatosis
WG is a rare systemic inflammatory disease of unknown etiology that has vasculitis as a major histologic manifestation. WG predominantly affects the upper and lower respiratory tract and the kidneys.7 Although this pattern of involvement is often referred to as the “classic triad” of WG, more frequently only one or two sites may be involved. In one series reported by DeRemee and associates, involvement of all three sites was seen in only 14 of 50 patients.8 “Limited WG” historically has been defined as disease involving the lungs without associated glomerular disease,9 although the term is also used to describe active disease without involvement that threatens the function of a vital organ or the patient’s life.10
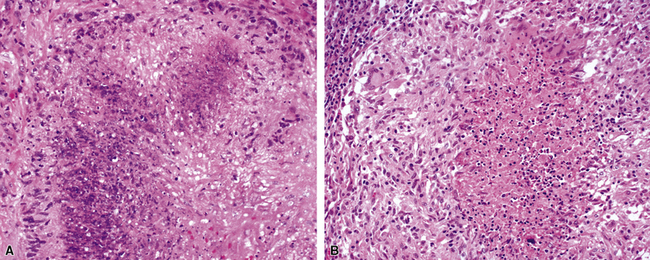
Despite the designation granulomatosis, well-defined granulomas without necrosis (sarcoid-like) are not a feature of this disease. The necrotizing lesions of WG have a peripheral zone of palisaded histiocytes, contrasting with the more epithelioid histiocytes seen at the periphery of necrosis produced by mycobacteria or fungi (Fig. 10-3). In fact, when well-formed (sarcoid-like) granulomas without necrosis are present in a potential case of WG, another diagnosis should be considered (usually infection).
Clinical Features
WG affects about 1 in every 3 million people in the United States11 and 1 in 8.5 million people in the United Kingdom.12 There is debate as to whether WG occurs more frequently during cold seasons, with some studies suggesting an increased occurrence in winter months.12 Other studies have disputed these results.11 Although the etiology of WG remains essentially unknown, several theories have been proposed. One theory is that an inciting inflammatory event invokes a specific immune response leading to the production of ANCAs (also discussed in the “Laboratory Studies” section), with ANCA playing a direct role in inciting tissue damage.13,14 A potential link between infections and the development of WG is being explored. It has been noted that a subtype of ANCA directed against lysosomal membrane–associated protein 2 (LAMP-2) is found in more than 90% of patients with pauci-immune necrotizing glomerulonephritis and frequently coexists with antiproteinase-3, and antimyeloperoxidase LAMP-2 has been observed to activate neutrophils, as well as causing injury to vascular endothelial cells in the absence of neutrophils. LAMP-2 cross-reacts with the bacterial adhesin FimH, and one study found that infection with bacteria expressing FimH occurred in 69% of patients in whom ANCA-positive glomerulonephritis subsequently developed.15,16 Although such findings suggest a potential link between infection and the production of ANCA, further study is needed in regard to a link between infection and WG. Similarly, other studies have demonstrated a link between T cells and the development of ANCA and have shown a particular role for T-helper 1 (Th1) lymphocytes, although the role of the Th1 lymphocytic pathway in the development of WG is still being elucidated.17,18 Genetic factors, toxic exposures, and deficient proteinase-3 clearance are also being explored as potential causes and contributing factors.11,19
WG occurs at any age but typically is a disease of adults, with a mean age of 50 years.20–22 A list of the clinical manifestations of WG is presented in Table 10-1. Body sites most commonly affected are the head and neck region, followed by the lung, kidney, and eye.20,23 Patients may experience a number of other complaints such as hoarseness, stridor, earache, hearing loss, otorrhea, cough, dyspnea, hemoptysis, or pleuritic pain. Pulmonary symptoms in the absence of upper respiratory tract manifestations are unusual. Destructive inflammation of the nose may result in a saddlenose deformity. In addition, patients may exhibit more generalized systemic signs and symptoms including arthralgias, fever, cutaneous lesions, weight loss, and peripheral neuropathy.24 Rarely WG may involve the salivary glands, pancreas, breast, mediastinum, gastrointestinal tract, prostate and urethra, vagina and cervix, heart, spleen, or peripheral or central nervous system.2,25–27
Table 10-1 Wegener Granulomatosis: Clinical Manifestations
| Manifestation | Frequency (%) | |
|---|---|---|
| At Presentation | During Course of Disease | |
| Head and neck manifestations | 73 | 92 |
| Sinusitis | 51 | 85 |
| Nasal disease | 36 | 68 |
| Otitis media | 25 | 44 |
| Hearing loss | 14 | 42 |
| Subglottic stenosis | 8 | 16 |
| Ear pain | 1 | 14 |
| Oral lesions | 3 | 10 |
| Pulmonary manifestations | 45 | 85 |
| Infiltrates | 23 | 66 |
| Nodule | 22 | 59 |
| Cough | 19 | 46 |
| Hemoptysis | 12 | 30 |
| Pleuritis | 10 | 28 |
| Renal manifestations | 18 | 77 |
| Eye manifestations | 15 | 52 |
| Conjunctivitis | 5 | 18 |
| Dacryocystitis | 1 | 18 |
| Scleritis | 6 | 16 |
| Proptosis | 2 | 15 |
| Eye pain | 3 | 11 |
| Visual loss | 0 | 8 |
| Retinal lesions | 0 | 4 |
| Corneal ulcers | 0 | 1 |
| Iritis | 0 | 2 |
| Systemic manifestations | ||
| Joints | 32 | 67 |
| Fever | 23 | 50 |
| Skin changes | 13 | 46 |
| Weight loss | 15 | 35 |
| Peripheral nervous system abnormalities | 1 | 15 |
| Central nervous system abnormalities | 1 | 8 |
| Pericarditis | 2 | 6 |
Data from Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener’s granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992;116:488–498.
Laboratory Studies
Nonspecific abnormalities on general laboratory tests are often present in patients with WG. The most common of these include leukocytosis, thrombocytosis (>400,000 cells/μL), marked elevation of the erythrocyte sedimentation rate, and normochromic normocytic anemia. In the past decade, the diagnosis of WG has been dramatically aided by the discovery and use of serum ANCA.28–32
Two major immunofluorescence patterns occur as expressions of ANCA (Fig. 10-4): the cytoplasmic or classic type (c-ANCA) and the perinuclear type (p-ANCA).33 The c-ANCA pattern is associated with WG and is present in the vast majority of patients with active generalized disease. Partial or complete remission of disease is reflected in a lower frequency of a positive test result, but 30% to 40% of patients in complete remission still have identifiable antibodies.34 The p-ANCA pattern can be seen in a small percentage of patients with WG, but it is more characteristic of idiopathic necrotizing and crescentic glomerulonephritis, microscopic polyangiitis, polyarteritis nodosa, and CSS.35
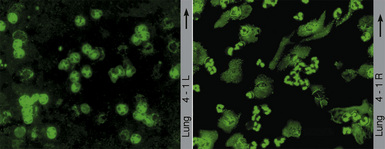
(From Travis WD, Colby TV, Koss MN, et al, eds. Non-Neoplastic Disorders of the Lower Respiratory Tract. In: King DW, ed. Atlas of Nontumor Pathology. Washington, DC: American Registry of Pathology and Armed Forces Institute of Pathology; 2002, Figure 4-1.)
The ANCA immunofluorescence patterns have been shown to correspond to specific antigen immunoreactivities; c-ANCA typically has specificity for proteinase-3, while most p-ANCAs have a specificity for myeloperoxidase. Studies have shown no significant difference in the lung biopsy findings from WG patients with c-ANCAs versus those with p-ANCAs.36,37 Levels of c-ANCA in the bronchoalveolar lavage fluid have not been shown to be a more specific predictor of WG or of the level of disease.38 Importantly, the presence or absence of a positive serum test for c-ANCA alone is not sufficiently specific to make or exclude the diagnosis of WG, and c-ANCA may occasionally be encountered in patients with other vasculitic syndromes or infection.39
Radiologic Features
Most patients with pulmonary disease have multiple opacities (Figs. 10-5 and 10-6) in the form of well-marginated nodules or masses of variable size (0.5–10 cm). Lesions may wax and wane over time. Most occur in the lower lobes.40–42 Poorly defined or even spiculated nodules may also be seen.43 Cavitation of nodules occurs in 25% to 50% of cases, with cavity walls typically being thick and irregular. Such lesions may evolve into thin-walled cysts or disappear completely with therapy.42,44 WG is often included in the differential diagnosis for interstitial lung disease because multifocal, ill-defined parenchymal consolidations can occur (with or without cavitation) and diffuse reticular and nodular interstitial opacities have also been reported.42,45
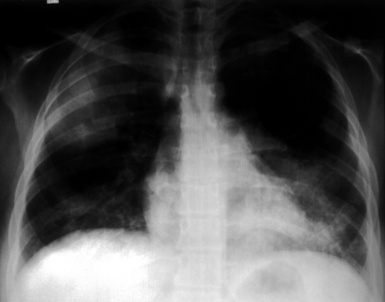
(From Travis WD, Colby TV, Koss MN, et al, eds. Non-Neoplastic Disorders of the Lower Respiratory Tract. In: King DW, ed. Atlas of Nontumor Pathology. Washington, DC: American Registry of Pathology and Armed Forces Institute of Pathology; 2002, Figure 4-2.)
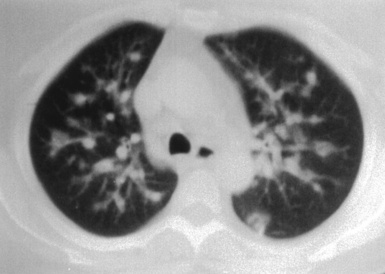
(From Travis WD, Colby TV, Koss MN, et al, eds. Non-Neoplastic Disorders of the Lower Respiratory Tract. In: King DW, ed. Atlas of Nontumor Pathology. Washington, DC: American Registry of Pathology and Armed Forces Institute of Pathology; 2002, Figure 4-3.)
Patients with WG may present initially with pulmonary hemorrhage. In this setting, diffuse infiltrates on chest radiographs and diffuse air space opacities on computed tomograms are observed (see Fig. 10-6). In children, pulmonary hemorrhage is a common presentation of WG, while pulmonary nodules occur less frequently in pediatric patients.22
Pleural effusion accompanies WG in 20% to 50% of cases, sometimes with focal pleural thickening. Hilar or mediastinal lymphadenopathy is an unusual finding in WG and, when significant, should raise concern for an alternate diagnosis. On rare occasions, WG occurs as a solitary pulmonary nodule (with or without cavitation), or as an isolated area of consolidation.46
Computed tomography (CT) provides optimal visualization of the number, location, and morphologic characteristics of the pulmonary abnormalities in WG. Well-marginated nodules and masses, sometimes with spiculated borders, are typical findings. A “feeding” vessel is seen in 88% of nodules (see Fig. 10-6), consistent with the angiocentric nature of this disorder.47 Cavitation is identified in 50% of cases. Another very common finding in WG is wedge-shaped peripheral opacities mimicking the CT appearance of infarct. Other, less common radiologic presentations include air bronchograms and the CT halo sign (ground glass opacity surrounding a pulmonary nodule or mass).47,48 Stenosis of the trachea or large airways may occur in short or long segments and may be complicated by partial or complete lobar collapse.45,49,50
Pathologic Features
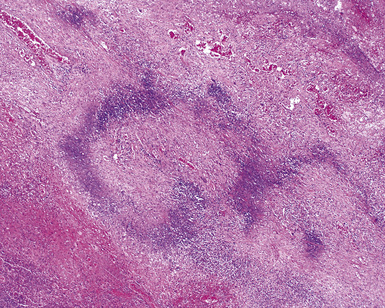
WG is characterized by the presence of multiple bilateral pulmonary nodules, often with cavitation51 (Fig. 10-7; see also Fig. 10-6). Solid nodular zones of consolidation with areas of punctate or geographic necrosis are typical findings (Figs. 10-8 and 10-9). WG can rarely present with a solitary lung lesion, but solitary granulomatous disease is more likely to be of infectious origin.52 When dealing with a solitary granulomatous lung nodule, a combination of both the classical histology and typical clinical or serologic findings of WG should be present before making a diagnosis.46 Even when special stains for organisms and cultures are negative, most of these solitary lesions represent old fungal or mycobacterial infection. Rarely the lesions of WG may predominantly involve bronchi. When acute lung hemorrhage is prominent, the cut surface of the lung is bloody and dark red.
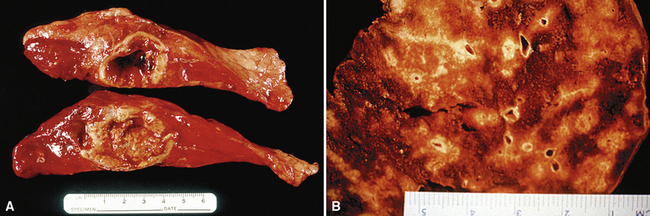
(From Travis WD, Colby TV, Koss MN, et al, eds. Non-Neoplastic Disorders of the Lower Respiratory Tract. In: King DW, ed. Atlas of Nontumor Pathology. Washington, DC: American Registry of Pathology and Armed Forces Institute of Pathology; 2002, Figure 4-5.)
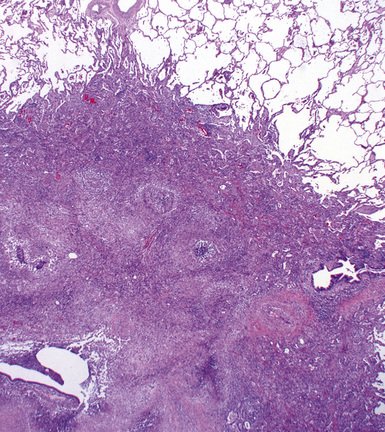
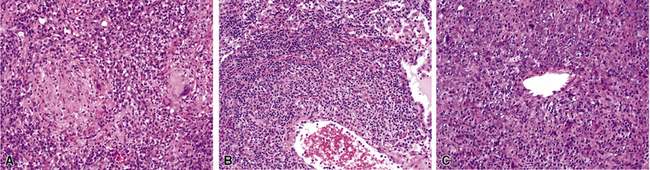
At scanning magnification, the pulmonary lesions of WG simulate their radiologic appearance (see Fig. 10-8). The classic findings consist of nodular areas of consolidation with variable zones of necrosis. Major diagnostic criteria, presented in Box 10-2, include parenchymal necrosis (see Fig. 10-9), vasculitis (Fig. 10-10), and granulomatous inflammation (Fig. 10-11). Another important feature is a mixed inflammatory infiltrate composed of neutrophils, lymphocytes, plasma cells, macrophages, giant cells, and eosinophils (Fig. 10-12). Parenchymal necrosis can take the form of neutrophilic microabscesses (Fig. 10-13) or large zones of geographic necrosis (see Fig. 10-9). The neutrophilic microabscesses are nearly pathognomonic of the disease and can be found within the mixed inflammatory infiltrate or within fibrous connective tissue including the adventitial collagen of larger arteries and veins and the pleura. Early microabscesses may consist of a small collection of neutrophils surrounding a focus of degenerated, often hypereosinophilic, collagen.51
Box 10-2 Wegener Granulomatosis: Major Histopathologic Manifestations (Diagnostic Criteria)
Data from Travis W, Koss M. Vasculitis. In: Dail D, Hammar S, eds. Pulmonary Pathology. New York: Springer-Verlag; 1994:1027–1095; and Travis WD, Hoffman GS, Leavitt RY, et al. Surgical pathology of the lung in Wegener’s granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol. 1991;15:315–333.
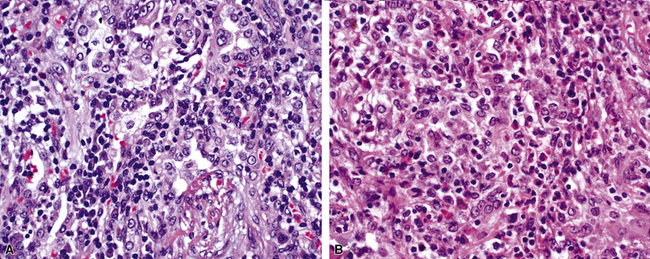
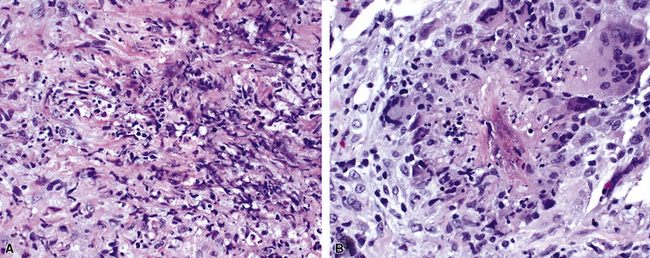
As illustrated in Figure 10-9, the classic geographic necrosis of WG is typically basophilic, owing to the presence of numerous necrotic neutrophils. The necrotic centers of WG lesions often lack the “ghosted” image of lung structure, a diagnostic clue useful in the case with atypical features (Fig. 10-14). This likely occurs because the necrotic zones of WG generally are not the result of “infarct-like” zonal parenchymal necrosis but rather occur by progressive expansion of collagen necrosis.
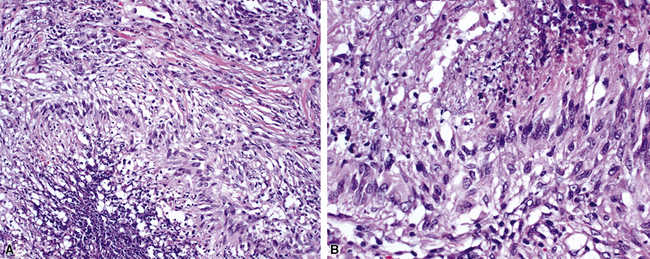
The “granulomatous” inflammation of WG typically includes giant cells scattered randomly or in loose aggregates.10–15 Also commonly observed are palisaded histiocytes (Fig. 10-15), giant cells lining the border of geographic necrosis or microabscesses, and microgranulomas consisting of small foci of palisaded histiocytes arranged in acartwheel pattern around a central nidus of necrosis51 (Fig. 10-16). The presence of tightly cohesive, sarcoid-like granulomas is very rare in WG and suggests infection or necrotizing sarcoid. Also, the presence of granulomas without associated necrosis favors an infectious etiology over WG.
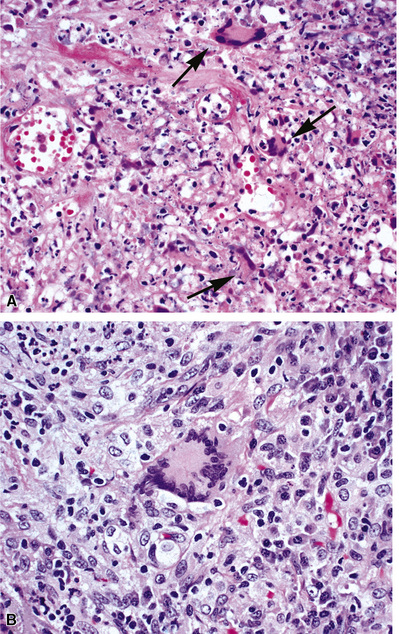
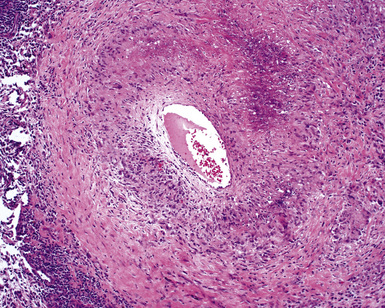
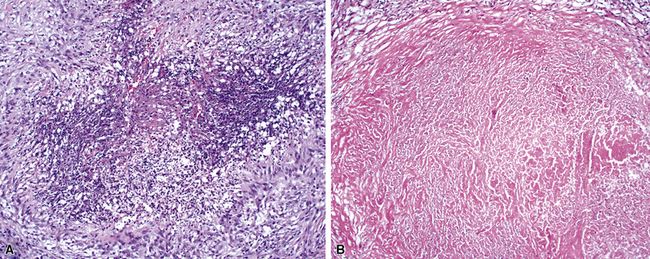
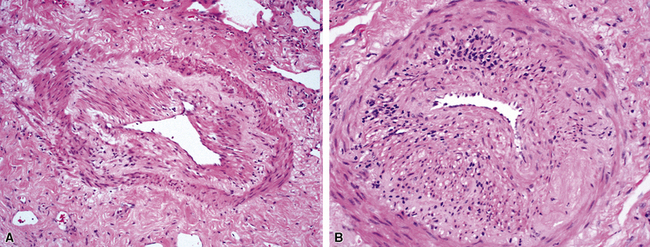
The vasculitis of WG typically affects small arteries and veins up to 5 mm in diameter. When vasculitis is seen in the surgical biopsy, it most often occurs within the dense inflammatory infiltrate surrounding nodular or geographic areas of necrosis (Fig. 10-17). Vasculitis in WG may comprise a variety of inflammatory cells including acute or chronic mural inflammation, necrotizing stellate granulomas, non-necrotizing stellate granulomas, and giant cells.51 Cicatricial changes consisting of mural fibrosis or luminal obliteration may be seen in specimens following therapy. Destruction of the vascular elastic laminae is commonly observed (Fig. 10-18). Sometimes the inflammation is limited to the endothelium (endotheliolitis) and subendothelial aspect of the vessel wall. Despite these potential vascular changes, if necrotizing vasculitis is held as a requirement for the diagnosis, many cases of WG will be missed.
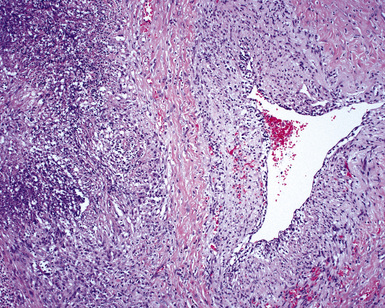
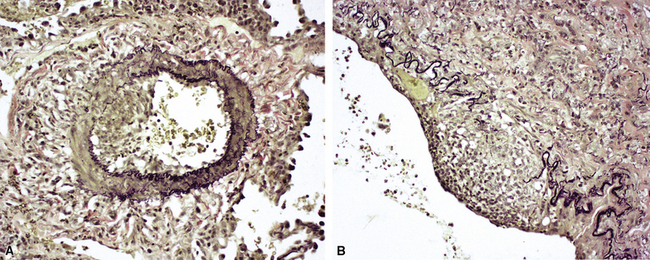
Another distinctive vascular manifestation of WG is capillaritis (Fig. 10-19). In many cases, capillaritis is only focally evident in the biopsy.51 When capillaritis is prominent, it is distinctive and easily recognized. In the rare case of WG dominated by capillaritis, a careful search throughout the rest of the biopsy should be made for more typical findings of WG such as granulomas, foci of necrosis (such as neutrophilic microabscesses), multinucleate giant cells, and vasculitis affecting arterioles or veins.
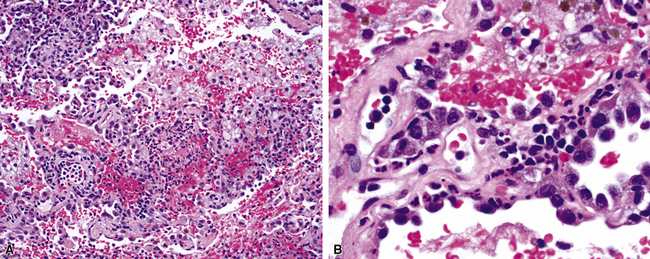
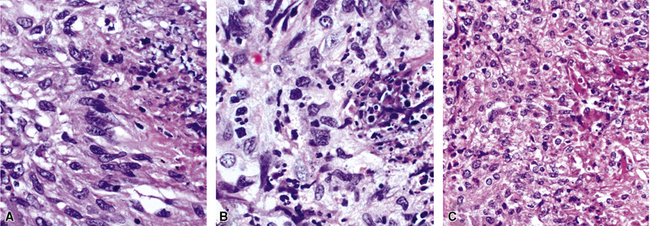
In addition to these major histologic features, a variety of minor histologic features may be encountered (Box 10-3), including alveolar hemorrhage, interstitial fibrosis, lipoid pneumonia, organizing pneumonia, lymphoid hyperplasia, extravascular tissue eosinophils, and xanthomatous lesions. WG can also involve the airways, causing chronic bronchiolitis, acute bronchiolitis or bronchopneumonia, the histologic pattern of organizing pneumonia (see further on), bronchocentric granulomatosis, follicular bronchiolitis, and bronchial stenosis.51,53 Occasionally one of these minor lesions may be the dominant lung biopsy finding.51 Diffuse pulmonary hemorrhage is a severe life-threatening manifestation of WG. The pattern of bronchocentric granulomatosis is another rare manifestation of WG encountered in 1% of cases.51,53 Organizing pneumonia (Fig. 10-20) can be seen in 70% of lung biopsies from patients with WG51; rarely, it may be sufficiently dominant that some have referred to this manifestation as the “bronchiolitis obliterans organizing pneumonia” (BOOP) variant of WG.51,54 This should not be confused with the idiopathic entity of BOOP (cryptogenic organizing pneumonia) but should be recognized as nonspecific secondary organization following alveolar injury related to the underlying lesions of WG.
Box 10-3 Wegener Granulomatosis: Minor Histopathologic Manifestations*
Data from Rose A, Sinclair-Smith C. Takayasu’s arteritis. A study of 16 autopsy cases. Arch Pathol Lab Med. 1980;104:231–237; and Jakob H, Volb R, Stangl G, et al. Surgical correction of a severely obstructed pulmonary artery bifurcation in Takayasu’s arteritis. Eur J Cardiothorac Surg. 1990;4:456–458.
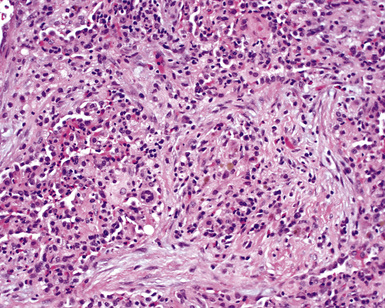
The lung biopsy findings from patients with WG may not show classical histologic findings, especially if patients are either biopsied very early in the course of disease or following therapy.51,55 Interstitial fibrosis (sometimes with scattered giant cells, but without necrosis)(Fig. 10-21), bronchial or bronchiolar scarring, and cicatricial vascular changes (Fig. 10-22) are common in lung biopsies from patients who have received therapy.51,55 Wedge biopsies provide the best results for an accurate diagnosis of WG. Transbronchial biopsies rarely yield diagnostic information, although in the appropriate clinical context the presence of a few neutrophil microabscesses, giant cells, or capillaritis may be helpful in supporting the diagnosis. Transthoracic needle core biopsies may occasionally show features suggesting a diagnosis of WG.
Differential Diagnosis
The differential diagnosis for WG based on lung biopsy tissue depends somewhat on the constellation of changes present and includes granulomatous infection,52 lymphomatoid granulomatosis,56,57 CSS,58–61 sarcoidosis, necrotizing sarcoid granulomatosis,56,62,63 rheumatoid nodules,63 bronchocentric granulomatosis,53,63,64 and diffuse pulmonary hemorrhage syndromes.65,66
Occasionally, a form of diffuse large B cell malignant lymphoma commonly referred to as lymphomatoid granulomatosis (see Chapter 15) can bear a striking resemblance to WG6 (Fig. 10-23). Like classic WG, this neoplastic process is characterized pathologically by the presence of multiple necrotic pulmonary nodules. In addition to major clinical differences between these diseases, important histopathologic differences become evident at closer inspection. First, the necrotic areas in lymphomatoid granulomatosis typically demonstrate pale shadows of large necrotic cells (dead lymphoma cells). Second, within the necrotic zones, and at the periphery of necrosis, medium-sized blood vessels can be seen whose outline is expanded by an angiocentric infiltration of lymphoid cells. As noted, this is a diffuse large B cell lymphoma in which the atypical B lymphoid cells are infected with Epstein-Barr virus (EBV) and associated with a T lymphocyte–rich inflammatory reaction and vasculitis. In high-grade disease, the vasocentric infiltrate is composed mainly of large atypical B cells. In lower-grade forms, the infiltrate may be polymorphous with more prominent T cells and a mixture of plasma cells, and eosinophils. Immunohistochemistry for CD20 and CD3 highlights the large malignant B cells and background of inflammatory T cells. Immunohistochemistry for EBV latent membrane protein 1 (LMP-1) and in situ hybridization studies for EBV are valuable diagnostic tools in this setting. Third, lymphomatoid granulomatosis is a vasodestructive lymphoid neoplasm, so necrosis and obliteration of vessels are common. WG may show necrosis in vascular adventitia, but wholesale medial necrosis in arteries and veins is unusual. Fourth, the atypical cells of lymphomatoid granulomatosis often contain EBV,4,67 a finding not expected in WG. Finally, granulomatous inflammation is comparatively rare in lymphomatoid granulomatosis, so the presence of granulomas in nodular lung lesions should suggest a diagnosis other than lymphomatoid granulomatosis (e.g., infection or WG).
Prominent tissue eosinophilia occurs in approximately 5% of cases of WG (Fig. 10-24). With this finding, the differential diagnosis should include CSS (see later), along with fungal or parasitic infection.58,61,68,69 Peripheral blood eosinophilia is characteristic of CSS and is uncommon in WG.70 Also, asthma is not a characteristic feature of WG, although rarely, asthmatic individuals may develop WG, presumably at a rate similar to that seen in the general population. The distinction between WG and CSS is usually straightforward, but some cases may require careful assessment of all of the clinical, pathologic, and laboratory data (Table 10-2).
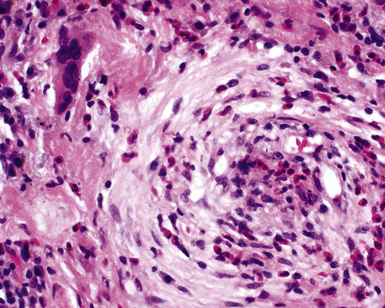
Table 10-2 Wegener Granulomatosis versus Churg-Strauss Syndrome: Distinguishing Features
| Clinical/Pathologic Feature | Wegener Granulomatosis | Churg-Strauss Syndrome |
|---|---|---|
| Asthma | Rare | Characteristic (diagnostic criterion) |
| Eosinophilia | ||
| Peripheral | Up to 12% | Characteristic* |
| Tissue | Up to 6% | Characteristic* |
| Sinus disease | Destructive, often causing saddlenose deformity | Less severe, usually allergic rhinitis |
| Renal disease | More severe | Usually mild |
| Cardiac disease | Rare | Common |
| ANCA | Usually c-ANCA | Usually p-ANCA |
ANCA (c-ANCA, p-ANCA), antineutrophil cytoplasmic antibodies (cytoplasmic, perinuclear).
* Eosinophilia may be fleeting and may be difficult to demonstrate during steroid therapy.
Perhaps the most important, and often problematic, consideration in the diagnosis of WG is the exclusion of infection. Mycobacteria and fungi can cause necrotizing granulomatous inflammation and vasculitis resembling that seen in WG. Solitary necrotizing granulomas can be associated with vasculitis in 87% of mycobacterial lung infections and 57% of fungal lung infections.52 Also, neutrophilic microabscesses are a feature of certain infections, such as blastomycosis and nocardiosis.
A number of important clues can be helpful in the approach to this differential diagnosis, even before special stains for organisms or culture data (which should be routinely ordered in such cases) are available. First, if the lesion is solitary, a high index of suspicion for infection is appropriate.46 Second, WG does not tend to make granulomas without central necrosis,51 except in the rare occurrence of infection superimposed on the necrotic center of a WG lesion. Third, the necrosis of infection may show the “ghosted” outlines of underlying lung parenchyma, a finding uncharacteristic of WG. Fourth, the patient with infection, in whom a bilateral multiple-nodular appearance on radiologic studies may simulate that in WG, is typically quite ill, with generalized systemic symptoms. By contrast, the patient with WG may be relatively asymptomatic, despite numerous necrotic nodules in the lung. Finally, when strictly morphologic assessment fails to clarify the diagnosis, inquiry regarding the presence of sinonasal disease or renal disease and serologic data (c-ANCA and p-ANCA) will usually resolve the quandary.
When WG presents with a predominantly bronchocentric pattern of lung involvement, bronchocentric granulomatosis must be considered in the differential diagnosis.51,53 Patients with bronchocentric WG should demonstrate other distinguishing features of WG, including renal or sinus involvement and a positive ANCA serology.
Diagnosis
The histologic features of WG can be very suggestive of the diagnosis, but as a general rule, it is extremely important to correlate the histopathology with clinical and serologic findings before making a definitive diagnosis on a lung biopsy specimen. The diagnosis can be impossible to make in cases in which only partial clinical or pathologic criteria are present. In these situations a purely descriptive diagnosis with a differential diagnosis may be necessary. As mentioned earlier, ANCA serology can be helpful, as long as one keeps in mind that ANCAs are not specific for WG.71 Moreover, when all other clinical and histopathologic findings are compelling for WG, the diagnosis is still possible despite negative ANCA studies.39
Treatment and Prognosis
WG is commonly a fatal disease if left untreated, with up to 90% of patients dying within 2 years of diagnosis, most often from respiratory or renal failure. Fortunately, therapy with cyclophosphamide and prednisone is very effective in achieving remissions, with 85% to 90% of patients responding to therapy and approximately 75% experiencing complete remission.72 The median time to remission is 12 months, although occasional patients require treatment for more than 2 years before all symptoms resolve. Even in those patients who initially respond to therapy, relapses are common, with up to 50 of initial responders experiencing at least one relapse requiring another course of therapy. Trimethoprim-sulfamethoxazole, pulse cyclophosphamide, and methotrexate are also used to treat WG.20,73–75 Trimethoprim-sulfamethoxazole may reduce relapses for those patients who are in remission.76 The mechanism of this protective action is unknown. Rituximab has more recently shown promise in treatment of WG, particularly limited disease refractory to standard therapy.77 Initial trials of antagonists of tumor necrosis factor-alpha (TNF-α) showed some benefit, but formal trials did not support the initial findings; thus, further study is needed to determine the efficacy of this potential therapy.78 The outcome with WG seems to be significantly worse for patients older than 60 years of age than for younger patients, despite similar clinical manifestations and treatment regimen. Lung function frequently improves after treatment, but in some patients the diffusing capacity may never return to normal.
Churg-Strauss Syndrome
CSS is a multisystem disorder characterized by the triad of asthma, peripheral blood eosinophilia, and vasculitis.58,59,61,68–70,79–81 Although CSS was initially described by Churg and Strauss based on a series of autopsy cases,58 it is now recognized primarily as a clinical entity. Accordingly, most cases today are diagnosed on the basis of clinical findings rather than lung biopsy.61
In 1990, the American College of Rheumatology (ACR) proposed two approaches to the diagnosis of CSS (Table 10-3): a traditional format classification and a classification tree.82,83 According to the traditional format classification, six criteria are identified: (1) asthma, (2) eosinophils greater than 10% of the white blood cell differential count, (3) mononeuropathy (including multiplex) or polyneuropathy, (4) non-fixed radiographic pulmonary infiltrates, (5) paranasal sinus abnormalities, and (6) a biopsy containing a blood vessel with extravascular eosinophils.82 If four of six of these criteria are met, the diagnosis can be established with a sensitivity of 85% and a specificity of 99.7%.82 The ACR criteria for CSS have been retained in the subsequent 1994 Chapel Hill consensus conference criteria and the more recently proposed Watts criteria.84
Table 10-3 Churg-Strauss Syndrome: Clinical Manifestations
| Manifestation | Frequency (% of Patients Affected) |
|---|---|
| Pulmonary infiltrates | 72 |
| Mononeuritis multiplex | 66 |
| Abdominal pain | 59 |
| Arthritis/arthralgias | 51 |
| Mild/moderate renal disease | 49 |
| Purpura | 48 |
| Cardiac failure | 47 |
| Myalgia | 41 |
| Löffler syndrome | 40 |
| Erythema/urticaria | 35 |
| Diarrhea | 33 |
| Pericarditis | 32 |
| Skin nodules | 30 |
| Pleural effusion | 29 |
| Hypertension | 29 |
| Central nervous system abnormalities | 27 |
| Gastrointestinal bleeding | 18 |
| Renal failure | 9 |
Modified from Lanham J, Churg J. Churg-Strauss syndrome. In: Churg A, Churg J, eds. Systemic Vasculitides. New York: Igaku-Shoin; 1991:101–120.
The major criteria used in the classification tree are asthma, eosinophilia with greater than 10% eosinophils, and a history of allergy.82 According to this method, patients with well-documented systemic vasculitis, but lacking a history of asthma, can be diagnosed with CSS if they have peripheral blood eosinophilia (>10% eosinophils) and a history of allergy other than drug sensitivity.82 This seems appropriate, since patients without asthma, but with a history of allergic disease, can develop CSS.85–87 Both classification methods appear to be useful in the diagnosis, with greater sensitivity provided by the classification tree and greater specificity by the traditional approach.82



