Fig. 3.1
The prevalence “of pulmonary hypertension in Armadale, Australia and surrounding regions, December 2009 (Reproduced with permission from [1])
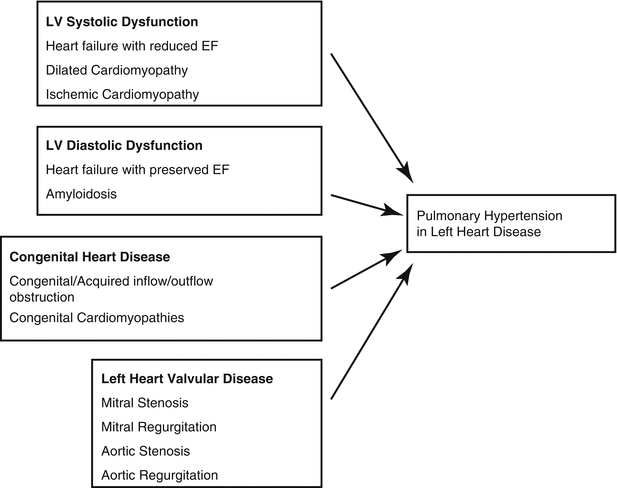
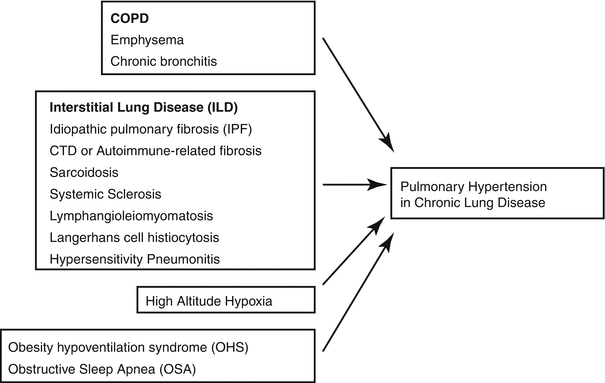
Fig. 3.3
Causes of Group 3 PH
Table 3.1
Comparison of Group 2 and 3 diagnostic findings
Group 2 PH | Group 3 PH | |
|---|---|---|
History | Dyspnea on exertion Lower extremity swelling | Dyspnea out of proportion to lung findings Nighttime awakenings, fatigue, morning headaches, snoring |
Risk factors | Rheumatic fever Hypertension OSA CAD, history of MI | Smoking Asbestos, Silica, Talc, Coal dust Autoimmune disease |
Examination | Elevated JVP Carotid bruits Irregular rhythm, murmur, S3 gallop, displaced PMI Crackles, dullness to percussion on lungs Hepatosplenomegally, ascites Lower extremity edema | Central obesity Barrel-chested Uveitis Pulmonary rales Clubbing of the extremities Erythema nodosum |
CXR | Cardiomegally Pleural effusions Pulmonary edema | Bilateral infiltrates Hyperinflation with flattened diaphragms |
Echocardiogram | Decreased EF Impaired relaxation Dilated, non-collapsible IVC | Mildly elevated RVSP Diastolic dysfunction |
PFTs | Restrictive pattern | Obstructive or restrictive patterns Decreased DLCO |
Pulmonary Hypertension Secondary to Left Sided Heart Disease
Epidemiology
Up to 250,000 patients in the United States have Group 2 PH, thereby making it much more prevalent than other causes of PH [7]. Approximately 60–70 % of patients with HFpEF and HFrEF develop PH [8] and up to half of patients referred to heart transplant clinics have pulmonary vascular resistance (PVR) greater than 2 Wood Units [9]. The development of PH in these patient populations is associated with a poor prognosis [10, 11]. In a study of HFrEF patients assessed for PH by echocardiogram, the 28 month mortality was almost 57 % in patients with PH compared to 17 % in HFrEF patients without PH [12]. Additionally, transplanted patients with PH have a greater risk of developing post-operative right ventricular failure and an increased risk of mortality [13]. In a community study of HFpEF patients, evidence of PH on echocardiography independently predicted survival [8]. However, treating patients with PH secondary to severe systolic heart failure with pulmonary vasodilators (prostaglandins) has caused pulmonary edema and death [13]. Thus, although PH predicts mortality, targeted PH treatments in PH-HFpEF may not improve outcomes, as will be subsequently discussed [4].
Pathophysiology
Elevated LV filling pressures lead to a passive increase in mPAP in order to maintain forward flow. Typically, this increase in mPAP is “proportionate” to the elevated LV filling pressure with a normal transpulmonary gradient and pulmonary vascular resistance (≤12 mmHg and ≤2.5 WU, respectively). This is referred to as “proportionate” or “passive” PVH. However, in some patients with left heart disease, the mPAP increases quite markedly in response to the elevated left sided filling pressure due to vasoconstriction or remodeling of the pulmonary arteries similar to PAH. This results in an elevated transpulmonary gradient (TPG) and PVR (>12 mmHg and >2.5 WU, respectively), referred to as mixed or reactive pulmonary venous hypertension. Mixed PVH can be either reversible with vasodilators when vasoconstriction is dominant or is fixed due to remodeling of the pulmonary arteries [14].
Recently, the summary documents from the 5th World Symposium on Pulmonary Hypertension discussed the forms of pulmonary hypertension in left sided heart disease. In this document, it was suggested that the term “out-of-proportion” PH should no longer be used in patients with severe post-capillary PH with increased left atrial pressure (LAP). Instead, increasing recognition was offered to the importance of describing the presence of a pre-capillary component in some cases of PH from left heart disease. In this setting, the authors proposed 2 types of PH from left heart disease, on the basis of the level of the diastolic pressure difference (DPD). DPD is defined as pulmonary artery (PA) diastolic pressure minus pulmonary capillary wedge pressure (PCWP). In this setting, “isolated post-capillary PH” is defined as: PCWP > 15 mmHg and DPD < 7 mmHg, and “combined post-capillary PH and pre-capillary PH” is defined as PCWP > 15 mmHg and DPD > 7 mmHg (Table 3.2) [4].
Table 3.2
Definition and classification of PH-LHD; LHD: Left heart disease
Terminology | PCWP (mmHg) | Diastolic PAP-PCWP (mmHg) |
|---|---|---|
Isolated post-capillary PH | >15 | <7 |
Combined post-capillary and pre-capillary PH | >15 | ≥7 |
In Group 2 PH, similar to Group 1, arteriolar remodeling is observed. Elevated left atrial pressure (LAP) is common to all forms of Group 2 PH. Backward transmission of this increase in LAP can mediate vascular changes. Acutely, an increase in LAP results in an increase in pulmonary venous pressure and, subsequently, pulmonary edema. Persistent increase in LAP produces fractures in the structure of the alveolar-capillary barrier by hydraulic forces [15], an event referred to as “capillary stress fracture” [16]. In this instance, the endothelial barrier is damaged and the extracellular matrix becomes weakened [17]. However, the pulmonary-capillary interface is typically able to restore its integrity with normalization of LAP [18]. With chronic elevation of LAP, the capillaries undergo vascular remodeling characterized by increased alveolar-capillary membrane thickness and increased deposition of collagen IV in the extracellular matrix (ECM) [19, 20]. This increase in membrane thickness can result in a decrease in diffusion capacity [15]. In HFpEF with PH there is pronounced medial hypertrophy in the small pulmonary arteries (PAs) along with the formation of a neointimal layer similar to what is observed in Group I PAH [21].
The variability in PA pressures in patients with heart failure (systolic or diastolic) has not been fully explained. There does appear to be an element of genetic variability. Serotonin (5HT) transporter polymorphisms have been found to influence the degree of increased PA pressure in patients with systolic heart failure [22]. This is relevant in light of the known vasoconstrictor properties of 5HT. In a study of patients with congestive heart failure (CHF), those patients who were homozygous for the LL genotype of the 5HT promoter had significantly higher mean pulmonary artery pressures (mPAP) compared to those CHF patients who were heterozygous for the promoter [22]. Interestingly, in patients with Group 2 PH secondary to mitral valve disease, decreased bone-morphogenetic protein (BMPR) expression has been reported, suggesting a common pathway to PAH in the development of PH in PVH [23].
Role of Nitric Oxide in Group 2 PH
Nitric oxide (NO) derived from the pulmonary vascular endothelial cells plays an important role in maintaining resting pulmonary vascular tone and pulmonary vasodilatation response to endothelium-mediated triggers [24, 25]. The constitutive enzyme, endothelial nitric oxide synthase (eNOS), in the pulmonary vascular endothelial cells produces NO from arginine. NO then diffuses from the endothelial cells to the neighboring pulmonary artery smooth muscle cells where it stimulates soluble guanyl cyclase, leading to increased cyclic guanosine monophosphate (cGMP). cGMP relaxes pulmonary artery smooth muscle by inhibiting the release of calcium from sarcoplasmic reticulum. Consistent with this, infusion of NG-monomethyl-L-arginine (L-NMMA), a non-specific inhibitor of eNOS, produces dose-dependent pulmonary vasoconstriction in normal adults and in children with congenital heart disease. In addition, cGMP reduces smooth muscle proliferation [26].
Experimental studies from animal models of chronic heart failure and chronic heart failure patients have shown reduced pulmonary endothelial cell derived NO in both populations [27]. Pulmonary arteries isolated from rats with chronic heart failure induced by ligation of the left coronary system showed reduced relaxation to acetylcholine, an eNOS agonist. Porter and colleagues studied the response of pulmonary arteries to acetylcholine infusion in heart failure patients with and without PH using intravascular ultrasound [28]. Patients with heart failure and PH had significantly reduced pulmonary vasodilatation in response to acetylcholine whereas those with normal pulmonary artery pressure had enhanced pulmonary vasodilatation. Similarly, Cooper and colleagues assessed pulmonary artery response to inhibition of NO with L-NMMA in CHF patients with and without elevated PVR by measuring segmental pulmonary arterial flow using Doppler wire [29, 30]. Taken together these studies demonstrate that PH due to left heart failure is associated with decreased pulmonary endothelium derived NO.
In addition to regulating vascular tone, endothelial-derived NO inhibits smooth muscle cell proliferation and thus decreased expression in heart failure begets more chronic changes in mPAP through vascular remodeling and platelet aggregation [31]. In light of this, strategies that increase pulmonary endothelial cell derived NO have attracted attention in the treatment of Group 2 PH.
Diagnosis
Diagnostic differentiation involves clinical features, echocardiogram assessment and invasive hemodynamics [21]. Risk factors for left ventricular dysfunction include increased age, hypertension, coronary artery disease (CAD), type 2 diabetes mellitus (DM2), obesity and atrial fibrillation [32, 33]. PH secondary to valvular disease or systolic heart failure can be determined through echocardiography relatively easily (Fig. 3.5). In contrast, it can be very difficult to distinguish between PAH and HFpEF. Assessing for clinical risk factors can accurately differentiate between PH secondary to HFpEF and PAH with more than 90 % accuracy [32]. Echocardiographic findings that favor a diagnosis of Group 2 rather than Group 1 PH include left atrial enlargement, concentric remodeling, left ventricular hypertrophy and evidence of impaired relaxation by Doppler measurements [34]. Opotowsky and colleagues developed an echocardiographic scoring system to distinguish between PH from HFpEF and PAH [35]. In this model, the presence of an E:e’ ratio of >10 (where E = mitral inflow velocities and e’ = mitral annular Doppler velocity) and left atrial dimension of greater than 4.2 cm had marked sensitivity for PH from HFpEF (Fig. 3.6).
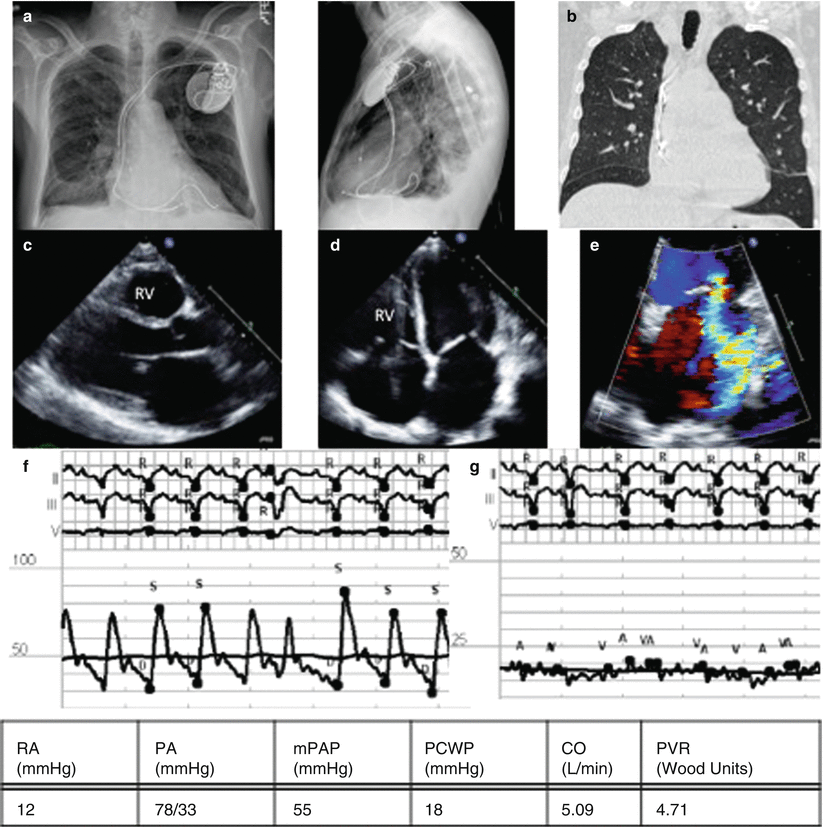
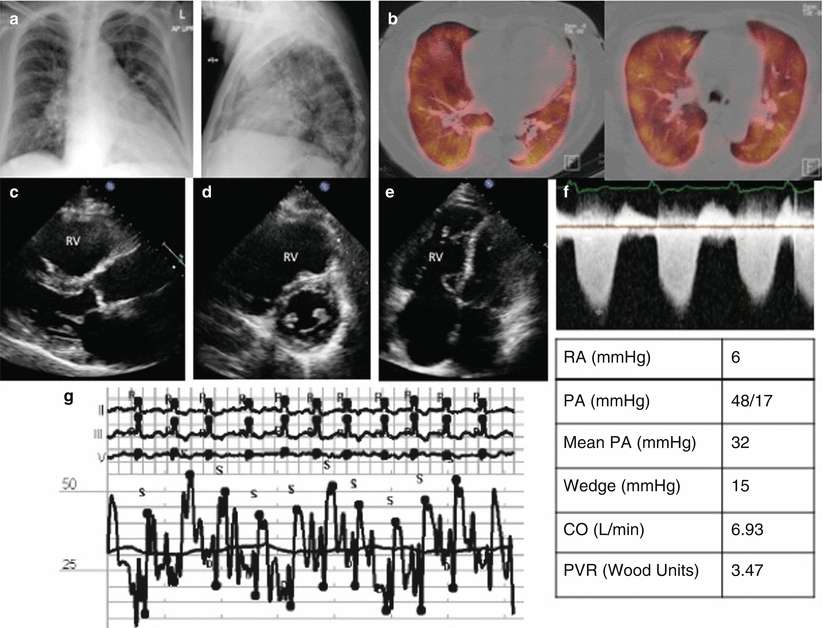
Fig. 3.5
Group 2 PH from left sided valvular disease – Mitral regurgitation. (a) CXR showing ICD, small pleural effusions, (b) Chest CT with apical septal thickening, a sign of pulmonary veno-occlusive disease, (c) Parasternal long axis, large LA, (d) Apical 4 chamber with large atria, (e) Color Doppler showing MR jet, RHC pressure tracings, (f) pulmonary artery pressures, mPAP 55, (g) PCWP tracing, PCWP 18
Fig. 3.6
Group 2 PH from heart failure with preserved ejection fraction. (a) PA/lateral CXR with cardiomegally and pulmonary edema, (b) Ventilation-Perfusion scan showing matched defects, (c) Parasternal long axis: large RV, (d) Parasternal short axis: large RV, (e) Apical 4 chamber: Large RV annexing the apex, (f) Tricuspid Doppler: TR velocity 359 cm/s, RVSP estimated at 52 mmHg, (g) RHC tracings of the pulmonary artery pressures, mPAP 32
Thenappan and colleagues demonstrated a mean PVR of 1.6 (1.3–2.0) Wood Units in HFpEF without PH, and a mean PVR of 4.8 (3.0–8.4) Wood Units in HFpEF with PH [32]. This range in PVR demonstrates that the PVR observed in some PH-HFpEF patients can rival that seen in PAH, thus underscoring the distinct pulmonary vascular disease (PVD) that can develop in these patients [32]. Left atrial enlargement, which had been successfully used to differentiate PAH from PVH, did not differentiate HFpEF from PH-HFpEF. However, right atrial enlargement was more commonly seen in PH-HFpEF than HFpEF, likely due to the increase in RV afterload secondary to the PVD [35]. PH-HFpEF patients have poorer functional capacity than HFpEF patients, reflective of the RV dysfunction observed. This finding highlights the need to distinguish these cohorts in clinical trials. Although hypertension, obesity and DM2 are known to increase the risk of developing diastolic dysfunction [36], none of these risk factors preferentially favored the development of PH-HFpEF over HFpEF alone or vice versa.
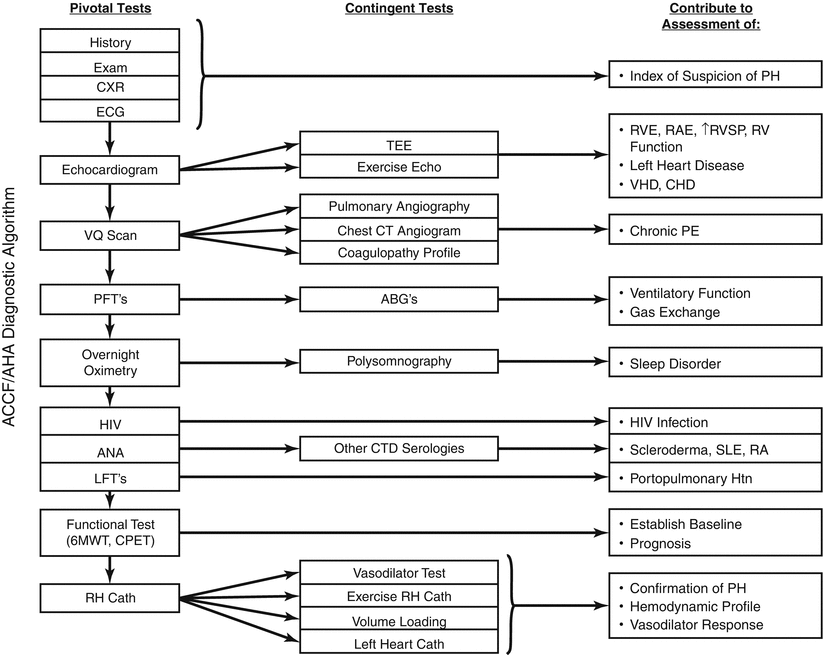
Despite advances in imaging, invasive hemodynamics are often required to definitively measure left sided filling pressures, through left ventricular end-diastolic pressure (LVEDP) or pulmonary capillary wedge pressure measurements (PCWP) [37]. When measuring PCWP, recording mean end-expiratory PCWP values is critical, as opposed to mean PCWP recorded by automated catheterization laboratory equipment, which may underestimate the left sided filling pressures in up to a third of patients [38].
In the setting of elevated pulmonary pressures, vasoreactivity testing with systemic and/or pulmonary vasodilator agents is used to assess if the PH is “fixed” in nature [39]. Currently, there is no standard internationally accepted protocol defining “fixed” or “reversible” PH or a consensus on the procedure to use in evaluation of PH. However, patients whose PVR decreases to less than 2.5 Wood Units with vasodilation in the setting of CHF, have improved outcomes at time of heart transplantation [9] Protocols to assess vasoreactivity include the use of nitroglycerin, nitroprusside, milrinone, among others [3], and vary by institution.
Resting catheterization is often insufficient to definitively rule in or out left ventricular dysfunction as the cause of PH. For example, at the time of catheterization left sided filling pressures can normalize secondary to diuresis [40]. Additionally, in early stages of HFpEF, neurohormonal activation is not as advanced resulting in less pronounced fluid retention. In these circumstances, right heart catheterization performed at rest and with exercise can distinguish between PH secondary to HFpEF (with increased PCWP at exercise) and exercise-induced PAH (which retains a normal PCWP) [41, 42]. In HFpEF, with exercise the increase in PA pressures associates closely with the increase in PCWP [3]. However, what defines an abnormal PCWP with exercise is not well described with most investigators suggesting a threshold of 15–25 mmHg. There is also uncertainty as to whether assessments should be performed supine or upright, with arms or legs exercising [33].
Alternative techniques to differentiate between Group 1 and Group 2 PH in the catheterization laboratory include rapid volume loading with saline infusion or leg lifting. A fluid challenge with 500 mL of normal saline over 5–10 min can identify patients with HFpEF and normal baseline PCWP, which may help reduce inappropriate diagnoses of PAH in patients with HFpEF [43, 44] However, even in healthy subjects a large volume fluid bolus can increased PCWP, thereby further confusing the role of this technique to distinguish between Group 1 PH and Group 2 PH secondary to HFpEF [45].
Management
The first step in managing Group 2 PH is treating the underlying disease process [4]. The priority is optimization of left sided filling pressures. Although pulmonary artery pressures can be expected to decrease significantly after appropriate treatment of the underlying etiology, targeted interventions may fail to remodel the pulmonary vasculature adequately to normalize pulmonary artery pressures.
In mitral stenosis, based on the American College of Cardiology (ACC)/American Heart Association (AHA) Valvular Heart Disease guidelines [46], percutaneous balloon mitral valvuloplasty (PBMV) is indicated for asymptomatic patients with moderate or severe MS and a pulmonary artery systolic pressure (PASP) > 50 mm Hg at rest or >60 mmHg with exercise. Exercise testing with hemodynamic assessment (Doppler or invasive) is recommended to evaluate the response of the mean mitral gradient and pulmonary artery pressure in patients with MS when there is a discrepancy between clinical symptoms or signs and resting echocardiographic findings [47]. An increase in RV systolic pressure to 60–70 mmHg should prompt consideration for valve surgery.
For patients with New York Heart Association class II-IV symptoms and mitral stenosis, surgical valve replacement should be pursued even if severe PH is present if the valve is not amenable to PBMV. Although PA pressures fall immediately after PBMV, in correlation with the reduction in mitral valve gradient [48], the maximal effect can be delayed up to 6 months after surgery [49].
In the setting of mitral regurgitation (MR), the 2014 ACC/AHA valve guidelines recommend earlier intervention for chronic MR [50] and in fact offer a Class IIa recommendation that mitral valve repair is reasonable for asymptomatic patients with chronic severe MR and resting pulmonary hypertension (PA systolic arterial pressure >50 mmHg). PH frequently complicates significant MR from flail leaflet being found in up to 25 % of patients [51]. When present, PH with MR is associated with major outcome implications, approximately doubling the risk of heart failure and death. Mitral valve surgery remains beneficial for these patients through either mitral valve repair or replacement.
Residual PH after mitral valve surgery is predictive of poor outcomes [52]. Although mitral valve repair or replacement is the optimum management of PH secondary to mitral valve disease, the rate and amount of regression of PH after mitral valve surgery varies considerably [53]. Patients with a decrease in left ventricular ejection fraction pre-operatively and fibroelastic deficiency as the underlying cause of mitral regurgitation are at increased risk of residual PH after mitral regurgitation surgery [54].
PH in aortic stenosis (AS) occurs secondary to diastolic dysfunction of the left ventricle and the accompanying left ventricular hypertrophy and elevated filling pressures [55]. In patients with severe symptomatic AS, about 50 % of patients have at least mild PH and 15–30 % of patients have severe PH with mean PAP > 50 mmHg [56–58]. PH is also more likely in AS if there is an accompanying decrease in LVEF and mitral regurgitation [59]. AS patients with severe PH have increased perioperative mortality in aortic valve replacement versus AS patients without PH. Furthermore, moderate to severe persistent pulmonary hypertension after aortic valve replacement for AS is associated with a decreased survival compared to normal pulmonary artery pressures or mild PH [60]. However, PA pressures do gradually decline post-operatively [61].
In the modern era of transcatheter aortic valve replacement (TAVR), the severity of PH before TAVR is associated with 2-year mortality rates of 14 %, 27 %, and 48 % for PA systolic pressures <30 mmHg, 30–60 mmHg, and >60 mmHg, respectively (p = 0.001). Patients who have persistently severe PH after TAVR have a worse prognosis than those whose PA systolic pressures decrease below 60 mmHg (2-year mortality rate: 50.0 % vs. 18.6 %; p = 0.001). PA systolic pressures can fall significantly within 90 days after TAVR [62], and by itself PH should not be a contraindication to valve replacement.
PH is found in 10–20 % of patients with aortic regurgitation (AR) [63] and occurs secondary to elevations in LVEDP. Unlike other valve lesions, historical reports suggest that there is no significant increase in operative mortality in patients undergoing aortic valve replacement (AVR) for AR with severe PH compared to those with mild PH or normal PA pressures [64]. Additionally, PA pressures decrease more predictably in patients after AVR for AR to near-normal levels as compared to AS [64].
Although treatment of advanced heart failure with decreased LVEF focuses on pharmacologic therapy, these patients are often considered for orthotopic heart transplantation. When PVR is greater than 2.5 Wood units and TPG is greater than 15 mmHg, mortality at 3 month and 1 year post-transplant is increased [65]. In those patients with a PVR <2.5 Wood Units, mortality at 1 year post-transplant is 5 % compared with 24 % in those with PVR >2.5 Wood Units [66]. Of note, in patients with a pre-transplant PVR > 2.5 Wood units in whom the PVR decreases to <2.5 Wood units with nitroprusside, mortality at 3 month post-transplant is considerably lower than those with “fixed” PH (3.8 % versus 40.6 %) [9]. Thus, fixed PH appears to portend a worse prognosis than reversible PH in heart transplant patients.
More recently, left ventricular assist device (LVAD) therapy has increased the options available for patients with systolic heart failure. In a retrospective study of 145 patients with systolic heart failure and severe PH treated by LVAD support, significant improvements were seen in mPAP, PVR and TPG after 6 months of LVAD support [67]. LVAD therapy for 6 months may be sufficient to adequately remodel the pulmonary vasculature and optimize patients for transplant. However, treatment beyond 6 months does not appear to have any additive effect on pulmonary vasculature [67]. There is perhaps an expanding role for the LVAD therapy a bridge to heart transplant in the management of patients with severe left ventricular systolic HF and PH [68].
Constriction and Restriction
Constrictive pericarditis is a pericardial compressive syndrome due to scarring, fibrosis, and pericardial calcification. The compressive phenomenon contributes to significant impairment of ventricular filling which is characterized by elevation of both left and right ventricular filling pressures and equalization of left and right ventricular diastolic pressures [69]. The causes of constrictive pericarditis include collagen vascular diseases (systemic lupus erythematosus), infection (tuberculosis) or as a consequence of trauma and traumatic hemorrhagic pericardial effusion. Treatment requires surgical removal of the pericardium, which should result in normalization of filling pressures and resolution of PH (Fig. 3.7) [70].
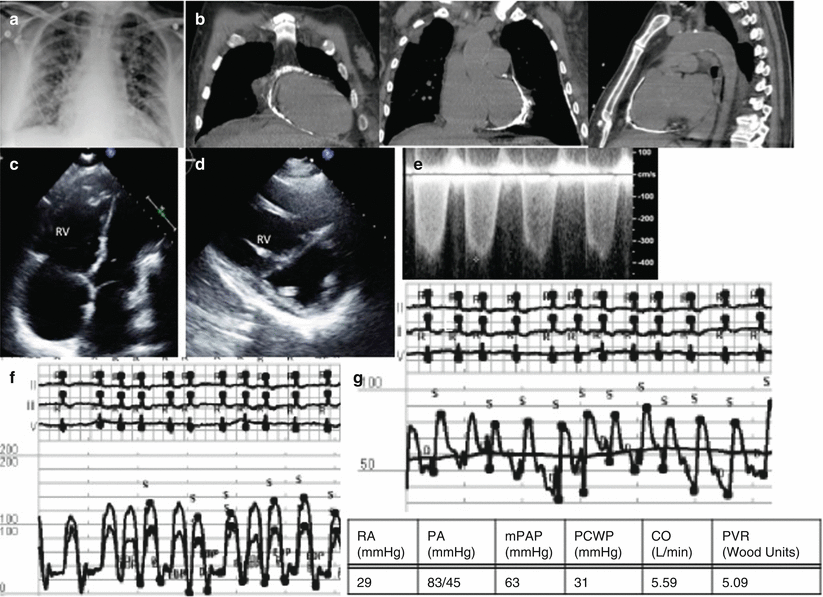
Fig. 3.7
Group 2 PH from constrictive pericarditis. (a) CXR with pulmonary edema and cardiomegally, (b) CT scan showing pericardial calcification, (c) Apical 4 chamber with septal flattening and RV bowing into the LV, (d) Parasternal short axis with septal bowing into the LV creating A “D” shaped LV, (e) TR velocity, RVSP estimated at 46.8 mmHg, (f) RV/LV tracing showing discordance of the ventricular pressure with inspiration, (g) RHC tracing of the pulmonary artery pressures, mPAP 63 mmHg
Restrictive cardiomyopathy is a myocardial disease characterized by decreased myocardial relaxation and increased myocardial stiffness, resulting in elevation of left ventricular end-diastolic pressures. The resultant increase in LAP contributes to postcapillary PH. The postcapillary PH can, in turn, cause an increase in right ventricular end-diastolic pressure, which can cause equalization of the diastolic pressures in restrictive cardiomyopathy. Amyloidosis is the most common cause of restrictive cardiomyopathy. Other causes are endomyocardial fibrosis, and eosinophilic myocarditis. Treatment is targeted at the underlying condition and in certain circumstances requires heart transplantation.
These conditions are best diagnosed with invasive hemodynamics. Simultaneous right and left ventricular pressure tracings will show a “dip-and-plateau” pattern in both these conditions and equalization of the diastolic pressures. Constrictive pericarditis is characterized by discordant change in RV and LV pressures seen with respiration. In restrictive physiology, the changes in RV and LV pressures are concordant with respiration (Fig. 3.7).
The Role of Pulmonary Vasodilators
Many of the currently available agents for PAH, namely, endothelin receptor antagonists (ERA), prostacyclins, phosphodiesterase 5 inhibitors (PDE5i), and soluble guanylate cyclase stimulators (sGCs), have been studied in Group 2 PH and have failed to demonstrate clinical benefit (Table 3.3) [3]. In the Flolan International Randomized Survival Trial (FIRST), patients with systolic heart failure (LVEF < 25 % and New York Heart Association (NYHA) IIIB or IV) were randomized to intravenous epoprostenol or conventional therapy. Although epoprostenol decreased pulmonary artery pressure and PVR, there was no observed benefit in morbidity or 6-min walk distance (6MWD). In fact, the trial was stopped early due to concerns for increased mortality in the epoprostenol arm [94]. The Endothelin Antagonist Bosentan for Lowering Cardiac Events in Heart Failure (ENABLE) study met with a similar fate [95]. In this study, 1,613 patients with LVEF < 35 % and NYHA IIIB or IV were randomized to bosentan or conventional therapy. In this trial, the results of which have never fully been published, there was an increased risk of heart failure hospitalization in the bosentan arm, presumably due to fluid retention or decreased right ventricular function [96].
Table 3.3
Pulmonary vasodilators studied in left heart failure
Study and year published | Methods | Inclusion criteria | PH at entry | Average mPAP at entry (mmHg) | Results |
|---|---|---|---|---|---|
Guazzi et al. [71] 2004 | Sildenafil 50 mg once or placebo n = 32 Randomized | >18 yrs NYHA II–III CHF LVEF < 40 % | No | mPAP = 33.6 ± 3.3 | Decreased mPAP Improved DLco Increased VO2 Improved ventilator efficiency |
Lewis et al. [72] 2007 | Sildenafil 25–75 mg tid or placebo 12-week study n = 34 Randomized | >18 yrs NYHA II-IV on conventional therapy LVEF < 40 % mPAP >25 mmHg | Yes | mPAP = 33 ± 3 | Decreased mPAP Improved peak VO2 Increased 6MWD Decreased hospitalization |
Guazzi et al. [73] 2007 | Sildenafil 50 mg bid or placebo 6-month study n = 46 Observational | <65 yrs NYHA II–III Cardiomyopathy LVEF <45 % | No | mPAP = 33.7 ± 3.1 | Decreased mPAP Improved peak VO2 Improved ventilator efficiency |
Lewis et al. [74] 2008 | Sildenafil 25–75 mg tid or placebo 12-week study n = 30 Randomized | >18 yrs NYHA II–IV LVEF < 40 % mPAP > 25 mmHg | Yes | mPAP = 30 ± 7 | Decreased mPAP Improved ventilator efficiency (decreased VE/VCO2 slope) Increased RVEF |
Behling et al. [75] 2008 | Sildenafil 50 mg tid or placebo 4-week study n = 19 Randomized | >20 yrs CHF on standard stable therapy LVEF < 45 % | No | mPAP = 59 ± 18 | Decreased mPAP Improved peak VO2 Improved ventilator efficiency |
Guazzi et al. [76] 2009 | Sildenafil 25 mg tid or placebo 6-month study n = 40 Observational | Age 65.3 ± 7.3 yrs LVEF 37.2 ± 7.4 % | No | Heart rate recovery improved | |
Guazzi et al. [77] 2011 | Sildenafil 50 mg tid or placebo 12-month study n = 44 Randomized | HFpEF LVEF > 50 % Sinus rhythm No hospitalizations in prior 6 mo | Yes | mPAP = 54.5 ± 6.3 | Decreased mPAP Improved RV function Decreased PVR Increased DLCO |
Guazzi et al. [78] 2011 | Sildenafil 50 mg tid or placebo 12-month study n = 45 Randomized | NYHA II–III LVEF < 40 % No hospitalizations in prior 6 mo Diabetic patients excluded | No | mPAP = 38.4 ± 3.0 | Improved LVEF Decreased PASP Improved LV diastolic function Improved functional capacity |
Guazzi et al. [79] 2012 | Sildenafil 50 mg tid or placebo 12-month study n = 32 Randomized | NYHA III–IV LVEF <45 %. mPAP 25–35 mmHg Females excluded | Yes | mPAP = 34.8 ± 4 | Reversal of exercise oscillatory breathing Improved functional capacity Decreased PAP |
Reichenbach et al. [80] 2012 | Sildenafil 20–60 mg tid or nothing 3-month study n = 47 Non-randomized | NYHA II–IV LVEF < 40 % | Yes | mPAP = 45 ± 6 | Decreased PVR Increased CO Increased TAPSE Decreased RV dimension |
RELAX [81] Ongoing | Sildenafil 20 mg tid for 12 weeks followed by Sildenafil 60 mg tid × 12 weeks n = 216 enrolled Randomized | >18 yrs NYHA II–III HFpEF, EF >50 % NT-proBNP > 400 pg/ml | No | 1° endpoint: Change in VO2 2° endpoint: 6MWD, composite clinical score | |
Givertz et al. [82] 2000 | Sitaxsentan IV 1.5, 3, 6 mg/kg over 15 min n = 48 Randomized | ≥21 yrs NYHA III–VI ACEI + diuretics PCWP ≥15 CI ≤2.5 | Yes | mPAP = 60 ± 2 | Decreased PASP, PVR, mPAP Unchanged CI, SVR |
HEAT Luscher et al. [83] 2002 | Darusentan 30, 100, 300 mg/day 3 week study n = 157 Randomized | NYHA III EF ≤35 % PCWP ≥ 12 CI ≤2.6 | No | Increased CI Unchanged PCWP, MPAP, PVR Unchanged BNP | |
Perez-Villa et al. [84] 2006 | Bosentan 62.5 mg BID × 4 weeks, then 125 mg BID 6 week study n = 7 Non-randomized | Heart transplant candidates PVR > 2.5 Woods units | Yes | mPAP = 48 ± 9 | Decreased PVR |
Kaluski et al. [85] 2008 | Bosentan 8–125 mg BID 5 month study n = 94 Randomized | NYHA IIIB–IV EF <35 % SPAP > 40 mmHg | Yes | PASP = 51.8 ± 10.3 | No change in sPAP Unchanged CI |
Hefke et al. [86] 2012 | Bosentan 62.5 mg BID up to 125 mg BID 12 month study n = 115 Observational, non-randomized | Heart transplant candidates mPAP > 35 PVR > 240 dyn TPG > 15 mmHg | Yes | mPAP = 45.2 ± 8.7 | Decreased PVR Decreased mPAP Decreased SVR Mortality much higher in placebo group |
Padeletti et al. [87] 2013 | Bosentan 62.5 mg BID for 1 month increased to 125 mg BID 4 month study n = 17 Non-randomized | >18 yrs NYHA III EF <35 % PASP >50, PVR > 3 Woods units, or TPG >10 mmHg | Yes | mPAP = 41 ± 7 | Decreased mPAP Decreased PVR Decreased TPR Unchanged CO |
Montalescot et al. [88] 1998 | Epoprostenol 4 ng/kg/min increase by 2 every 5 min to reach 15 % decreased in SBP n = 19 | >18 years Heart transplant candidates PASP >30 | Yes | mPAP = 41 ± 2 | Decreased mPAP Increased CO Increased PVC |
Weston et al. [89] 2001 | Inhaled Iloprost 580 μg single dose n = 6 Non-randomized, Observational | Heart transplant candidates mPAP > 25 Resistant to Nitroprusside | Yes | mPAP = 40.6 ± 10.9 | Decreased PASP Decreased MPAP Decreased TPG |
Sablotzki et al. [90] 2002 | Inhaled Iloprost 50 μg n = 29 (all male) Non-randomized, Observational | Heart transplant candidates Elevated PVR | Yes | PVR = 206 ± 94 | Decreased mPAP, PVR Increased CI, SI |
PROPHET [91] Scheidt et al. 2006 | IV PGE1 50 ng/kg/min up until PVR decreased n = 92 | Heart transplant candidates PVR >2.5 Woods Units TPG >12 mmHg | Yes | mPAP = 39 ± 9 PVR = 4.1 ± 2 | Decreased MPAP, SVR, PVR Increased CO |
Serra et al. [92] 2011 | IV PEG1 2.5 ng/kg/min up to optimal dose, continuously for 72 h n = 22 | NYHA III–IV EF <35 % ECHO-sPAP > 3 m/s | Yes | PASP = 57.65 ± 3.4 | Increased EF, NYHA class and PASP Improved 3 year mortality 27 % vs. 44 % |
LEPHT [93] Bonderman et al. 2013 | Riociguat 1 mg TID, 2 mg TID 16 week study n = 201 Randomized | >18 yrs mPAP ≥25 EF ≤40 % Optimal medical therapy × 30 days | Yes | mPAP = 40.4 ± 1.2 | Unchanged mPAP Increased CI, SVI Decreased PVR Improved quality of life |
PDE5i have a less clear effect in Group 2 PH. Sildenafil had been shown to improve 6MWD and decrease hospitalization in a small trial of 34 patients with systolic heart failure and PH [72]. In this study, there was not a significant reduction in pulmonary artery pressures, despite the observed clinical benefit [72]. Larger trials of PDE5i have yet to be completed [97] and a trial of tadalafil in Group 2 PH has recently been terminated by the funding agency [98]. Therefore the role of PDE5i in systolic heart failure and Group 2 PH currently remains unanswered. sGCs also act through the nitric oxide pathway on the pulmonary vasculature in a similar mechanism to PDE5i. Riociguat, currently the only available sGCs for PAH in the United States, has been studied in a phase IIb trial in systolic heart failure [93]. In this study, after 16 weeks of therapy, there was no observed change in pulmonary artery pressures.
The role of pulmonary vasodilators in Group 2 PH from HFpEF is also understudied. In a placebo-controlled study of 44 patients with Group 2 PH secondary to HFpEF, sildenafil improved exercise capacity and decreased pulmonary artery pressures after 6 months of therapy [77]. However, in a larger, multicenter study of 216 patients with HFpEF (median PASP was 41 mmHg), after 24 weeks of sildenafil versus placebo, there was no change in peak VO2, the study’s primary endpoint [99]. Admittedly in this trial, patients were not stratified by PH status, therefore the effect of this therapy on the pulmonary vasculature is as such undefined [100].
Future Directions
There remains considerable interest in using pulmonary vasodilators in the treatment of Group 2 PH. Clinical trials are ongoing and hopefully will provide insight into the merit of targeting pulmonary artery pressures [101, 102]. However, the lack of clinical benefit despite rigorous attempts to lower pulmonary artery pressures in Group 2 PH challenges the relevance of targeting PH in HFrEF and HFpEF [100].
Technological advances now permit the measurement of ambulatory hemodynamics with implantable monitors. For example, the CardioMEMS™ device is implanted into the branch pulmonary arteries and allows transmission of pulmonary artery pressure measurements, through a radiofrequency-based wireless pressure sensor. In the randomized, single-blind CHAMPION trial of 550 patients with NYHA functional class III heart failure, clinicians were directed to use a medical algorithm to achieve protocol specific hemodynamic targets: PASP 15–35 mmHg, Pulmonary Artery Diastolic Pressure (PAD) 8–20 mmHg, and mean PAP 10–25 mmHg. The CardioMEMS arm was associated with a 28 % relative risk reduction in HF hospitalization over the 6-month study period. In patients with PH who were managed with the CardioMEMS™ device, clinicians made more changes to HF medications, primarily decongestive in nature (diuretics, nitrates, vasodilators etc.) [103]. The accuracy of pressure measurement obtained was comparable to that obtained invasively with right heart catheterization [104]. Wearable technology as well as indwelling, continuous monitoring such as that performed by the CardioMEMS™ device, present a unique opportunity to characterize hemodynamics in patients when engaged in activities of daily living and will hopefully provide insight on optimal treatment practices.
Pulmonary Hypertension Secondary to Chronic Hypoxic Lung Disease
Chronic hypoxic lung disease commonly causes pulmonary hypertension (PH) with a variety of phenotypes. Among hospitalized patients with chronic lung disease, up to 28 % have exhibited elevated mean pulmonary arterial pressures (mPAP) [105]. This patient cohort is ever expanding with an aging, overweight population that smoked tobacco and had significant pulmonary exposures. It is important to recognize the lesser-known complications of chronic lung disease for prognostication and focused treatment.
Epidemiology
Chronic Obstructive Pulmonary Disease (COPD)
COPD often leads to an elevation of pulmonary pressures, affecting 30–70 % of patients [106]. These patients typically have advanced stage COPD, with mild to moderate elevation of their mean pulmonary arterial pressures (mPAP). However, there is a small population (5 %) of COPD patients that have severe PH. This cohort typically has mPAP >40 mmHg, with less severe airflow obstruction and hypoxemia [21, 107].
In emphysematous type COPD, worsening PH correlates with worsening airflow obstruction. Minai and colleagues found that 38 % of patients with severe emphysema had PH, 69 % of those being pre-capillary PH. It is important to note that COPD patients may show evidence of Group 2 PH as well, given that smoking is a strong risk factor for CAD [108]. Chronic bronchitis pattern COPD has been less thoroughly studied, although there is some data showing reversal of PH in chronic bronchitis when treated with oxygen therapy [109].
Interstitial Lung Disease (ILD)
The prevalence of PH ranges widely in patients with ILD and current prevalence is based on data primarily from patients referred for lung transplantation who had right heart catheterization (RHC). Idiopathic pulmonary fibrosis (IPF) is the most common form of ILD and as such has been studied most thoroughly. Rates of PH in IPF range from 31 to 84 %, depending upon the population studied [112, 113].
Prognostically, mPAP and FVC are the strongest predictors of survival in IPF [5]. A small subset of individuals with combined pulmonary fibrosis and emphysema are especially prone to PH and have high short term mortality with median survival ranging from 2.1 to 8.5 years [114]. In this cohort, the severity of PH and DLCO reduction do not correlate with the severity of obstructive lung disease [5]. Overall, in regards to lung disease, IPF carries a poor prognosis, with a median survival of 4.1 years and concomitant PH worsens that prognosis to a median survival of 0.7 years [115, 116].
ILD related PH also occurs in patients with pulmonary manifestations of connective tissue diseases, autoimmune disease and sarcoidosis. Similar to COPD, ILD related PH rarely exceeds the mild to moderate range with most mPAP < 40 mmHg [117]. In sarcoidosis the prevalence of pulmonary hypertension is extremely wide (5–75 %) with the latter finding in lung transplant candidates [118, 119]. Sarcoidosis manifests in many ways resulting in PH through different mechanisms that will be discussed subsequently.
Other autoimmune diseases typically included in the Group 1 PH category can manifest as Group 3 PH as well. In scleroderma-associated ILD, for instance, the incidence of PH has been estimated to be up to 22 % and a low PaO2 has been shown to correlate with development of PH more strongly than extent of lung fibrosis [120–123]. The prevalence of PH varies widely in all forms of ILD and there can be overlap of Group 1, 2 and 3 PH among the connective tissue disease and autoimmune patient populations.
High Altitude Pulmonary Hypertension (HAPH)
Increasing elevation portends a decrease in barometric pressure and concomitantly a decrease in the partial pressure of oxygen, most notably observed in patients at altitudes of 2,100 meters or greater above sea level. This atmospheric change manifests as worsened arterial oxygen saturation of hemoglobin with increasing altitude. The human body has multiple adaptation mechanisms to survive at altitude, which manifest as initial increases in cardiac output, followed by increase in hemoglobin production and decrease in plasma volume [124]. For the more than 140 million people living above 2,500 meters, this relative hypoxia can have significant health consequences, including PH. The primary geographic areas of the high altitude populations studied include the Himalayas, the Andes and the Rocky Mountains. Unfortunately, in a large number of these studies the mode of identifying patients with possible HAPH is EKG evidence of right heart strain and few of these patients undergo RHC. Studies of South American patients living at greater than 3,200 meters elevation have estimated prevalence of PH between 5 and 18 %; a few studies have shown higher pulmonary pressures in children than adults, especially those under the age of 5 [125, 126]. In a study of a Kyrgyz population, 14 % of adults were found to have EKG findings of right heart strain. A separate group of 136 Kyrgyz patients who underwent RHC showed that 20 % met criteria for PH [127].
There are many variables that influence the development of HAPH including elevation, sex, age and genetic susceptibility. HAPH is more common in men than women and in patients with OSA. The incidence also increases with higher elevation and older age as well. The prognosis of HAPH is favorable with removal of the stimulus, high altitude hypoxia, with a return of mPAP to normal values to be expected by 2 years at sea level [128].
Obesity-Hypoventilation Syndrome (OHS) and Obstructive Sleep Apnea (OSA)
Increasing rates of obesity have produced an increase in the number of patients with two, often overlapping, medical conditions that are associated with pulmonary hypertension: obesity-hypoventilation syndrome (OHS) and obstructive sleep apnea (OSA). Studies of patients with OHS on treatment with non-invasive positive pressure ventilation (NIPPV) have shown rates of PH up to 43 % by RHC, in which DLCO was lower and body mass index (BMI) was higher in the patients with elevated pulmonary pressures [129]. Previous studies of OHS patients not treated with NIPPV had shown a prevalence of 59 %, although it is important to note that both studies used mPAP > 20 mmHg diagnostic criteria for PH. PH is more common in OHS patients, as compared to purely OSA patients [130]. Minai and colleagues demonstrated that among patients with OSA, women have a higher rate of PH (86 % vs. 58 %) and those with Group 2 PH have higher frequency and length of nocturnal desaturations, as compared to those with Group 2 PH or normal mPAP [131]. The 8-year survival for patients with OSA and PH is significantly worse than those without PH (43 % vs. 76 %) [131]. PH in this setting will often improve with treatment of the underlying disease. Exercise capacity is also decreased when PH was present with OSA [131]. RV failure typically occurs in these patients when co-morbidities are present, such as left heart disease or hypoxic lung disease [132].
Pathophysiology
The mechanisms by which chronic lung disease causes PH are as diverse as the primary diseases themselves. Hypoxia results in distal changes in the pulmonary arterial system, initially vasoconstriction and, over time, muscularization of the vasculature [21, 133]. In large proximal arteries, medial and adventitial hypertrophy is predominantly seen [133].
The basis of much of the data on vascular remodeling in hypoxia is from examination of those individuals with high altitude pulmonary hypertension. In this population there is a production of α-smooth muscle actin (α-SM-actin) in the small pulmonary arteries that typically have very little smooth muscle, creating distal extension of the smooth muscle [133]. The protein α-SM-actin is a part of the contractile apparatus of smooth muscle cells and is a common marker of myofibroblast differentiation and increased fibroblast contractile activity [134, 135]. Medial and adventitial thickening appears to be due to this smooth muscle proliferation and extracellular matrix deposition. The adventitia also exhibits increased cellularity as a result of recruitment of fibroblasts. Initially, there seems to be an overall decrease in the size of the pulmonary vascular bed due to loss of small vessels in chronic hypoxia, however, animal model research has illustrated the presence of pulmonary angiogenesis in an attempt to decrease resistance across the vascular bed [136].
Hypoxia-Inducible Factor Alpha
One mediator of the adaptive response to hypoxia is hypoxia-inducible factor-alpha (HIF-α). There are two forms, HIF-1α and HIF-2α, the former activates glycolysis by upregulating Glucose transporter-1 (Glut-1) and the latter initiates erythropoietin (EPO) [137]. Glut-1 induces a shift to glycolytic metabolism in pulmonary vascular cells that correlates with a pseudo-hypoxic environment in the setting of inappropriate HIF-α activation [138, 139]. In the setting of normoxia, HIF-1α is bound to von Hippel Lindau (VHL) tumor suppressor protein (pVHL) and, when bound, ubiquinated HIF-1α cannot interact with DNA to alter transcription [140]. HIF-1α reacts to hypoxia by binding hypoxia response elements (HREs) in the DNA for molecules like vascular endothelial growth factor (VEGF) and erythropoietin (EPO) [141]. Interestingly patients affected by Chuvash syndrome, characterized by the developmental of polycythemia and PH, have a pVHL mutation that destabilizes the relationship of pVHL-HIF-1α in normoxia. In this disease, one mechanism of pulmonary vascular dysfunction is secondary to a perceived chronic hypoxic response in the absence of hypoxia. In mice, the development of PH appears to be independent of polycythemia [140, 142, 143].
Erythrocytosis does appear to play an important role in the development of PH, even outside of the Chuvash patient population. In COPD patients with polycythemia, phlebotomy correlated with a decrease in PVR and mPAP [143]. Increased blood viscosity is the mechanism by which erythrocytosis exacerbates PH, but does not appear to be the primary cause of elevated pulmonary pressures. Excess hemoglobin in erythrocytosis is thought to exacerbate hypoxic vasoconstriction by binding and removing large amounts of NO from the pulmonary circulation [142, 143, 145].
Microcirculation
In PH secondary to IPF, pulmonary vessel density is not uniform across parts of the lungs. The vessel distribution is thought to be secondary to an imbalance of angiogenic (VEGF and interleukin (IL)-8) and angiostatic (pigment epithelium-derived factor, PEDF) molecules [146–148]. This finding may be partially explained by the fact that VEGF transcription is induced by HIF-2α [149]. VEGF is involved in NO and prostacyclin synthesis in the lung, which leads to vasodilatation [150]. On a cellular level, VEGF does not appear to have significant mitogen effects on smooth muscle cells, but does stimulate transcription of matrix metalloproteinases-1,-3 and −9, that contribute to smooth muscle cell migration and digestion of the basement membrane as a first step in angiogenesis [151]. Inhibition of VEGF also correlates with endothelial cell apoptosis and elevated pulmonary pressures. Farkas and colleagues showed that vascular endothelial cell apoptosis, with microvascular rarefaction and pulmonary arterial medial thickening, correlated with decreased levels of VEGF in worsening IPF [149]. This reinforces the importance of anti-apoptotic properties of VEGF in maintaining lower pulmonary pressures, as represented by a negative correlation between mPAP and VEGF levels [149]. In the setting of ILD, it has been found that higher endogenous VEGF levels correlate with higher levels of fibrosis and poorer lung function [152]. IL-8 receptor (IL-8R) binding induces chemo-attractant molecule production and recruitment of neutrophils and other inflammatory cells. In animal models, overexpression of IL-8R has been shown to be protective by reducing the inflammatory response [153]. In rat models of PAH, transfusion of endothelial cells overexpressing IL-8R accelerates adherence to the injured lung and inhibits subsequent remodeling, likely by blocking inflammatory cell recruitment [154]. In IPF, higher levels of IL-8 are also predictive of increased mortality [155]. IL-8 is upregulated by hypoxia and is elevated in patients with COPD, correlating with disease severity [156]. PEDF is also instrumental in the remodeling of the lung endothelium and has a role in regulating angiogenesis as an angiostatic factor. PEDF is an inhibitor of VEGF angiogenesis and inhibits endothelial cell apoptosis and fibroblast growth. This activity may contribute to the development of PH in patients with IPF and emphysema, given that PEDF is elevated in these populations [147, 149, 157].
VEGF has been shown to play an important role, not only lung in vasculature, but also lung structure maintenance. This molecule likely participates in emphysematous changes and it has been hypothesized that decreased expression of VEGF and VEGF Receptor-2 in emphysema leads to alveolar septal endothelial apoptosis as a mechanism for wall destruction [158]. Additionally, chronic inhibition of VEGF in rats has been shown to lead to emphysema and that the addition of caspase inhibitors to halt apoptosis results in prevention of emphysema. Reduced levels of VEGF and the release of elastase by activated macrophages are two of the mechanisms known to produce alveolar septal destruction in emphysema [158].
Inflammation
In addition to VEGF, endothelial cell markers such as vascular cell adhesion molecule 1 (VCAM-1) and other vascular remodeling molecules are a topic of recent research. VCAM-1 is an adhesion molecule upregulated in hypoxia that facilitates inflammatory cell migration. A number of pro-inflammatory cytokines and growth factors are similarly upregulated in hypoxia, including monocyte chemoattractant protein-1 (MCP-1), IL-6, Transforming Growth Factor (TGF)-β1 and C5, with correlating increases in their respective receptors. Persistent hypoxia in animals models results in a predominantly monocytic perivascular infiltrate [159]. The recruitment of mesenchymal precursors of the monocyte line is profound and the corresponding influx of cells that produce type 1 collagen and α-SM-actin contributes to the expansion of the extracellular matrix (ECM) in vascular remodeling [160]. The increase in ECM thickens the vascular wall, thus increasing the distance of capillary from the alveoli thereby producing a paradoxical decrease in DLCO, worsening the hypoxic response [161].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


