Fig. 18.1
Right ventricular pressure–volume curve from normal subject (a) and from patient with increased right ventricular afterload (b). Adapted from Redington et al. [7]
Coronary perfusion also differs between the two ventricles. Myocardial perfusion in the LV occurs predominantly during diastole, when intra-myocardial tissue pressure falls below aortic root pressure. Because RV intra-myocardial tissue pressure remains below aortic root pressure throughout the cardiac cycle under normal loading conditions, the RV receives continuous coronary flow from the right coronary artery (RCA) [8, 9].
Last, the RV and LV cannot be viewed as isolated chambers because they share the same visceral cavity (the pericardium), common myofibers, and the interventricular septum. As a result of this ventricular interdependence and the contractile relationships noted above, RV systolic function depends significantly on the LV and changes in the condition of one ventricle can significantly impact the function of the other. Importantly, since both ventricles share myofibers in the septum, LV systolic function augments RV pressure generation and RV systolic performance [10, 11]. With these normal anatomical and physiologic characteristics in mind, the pathophysiologic derangements that occur in the ICU setting may be better understood.
Pathophysiology of RV Failure
Right ventricular failure can be defined as low CO and systemic hypoperfusion despite high RV filling pressures [12]. Because the RV is very sensitive to increases in afterload, rapid elevations in RV afterload decrease RV ejection fraction and induce RV dilatation [13]. In contrast, when RV afterload rises more gradually, adaptive RV myocardial hypertrophy may occur, reducing wall stress to maintain adequate stroke volume [14–18]. In the acute setting, a pressure-overloaded RV dilates and RV end-diastolic volumes and pressures rise, increasing RV wall stress and placing the RV on the descending portion of the Frank-Starling curve [19]. The Laplace relation, which states that wall stress is inversely proportional to the thickness of the wall, helps explain why the thinner RV free wall experiences a greater rise in wall tension with incremental elevations in RV pressure compared with the thicker LV free wall [8].
Higher wall stress in the RV increases myocardial oxygen demand and oxygen consumption [20, 21]. As RV wall tension increases, RCA blood flow, normally continuous during both systole and diastole, occurs only during diastole, causing a further decrease in oxygen delivery to the RV [9, 22]. The combination of increased myocardial oxygen demand and decreased oxygen supply leads to RV ischemia and decreased RV contractility [20, 23]. Right ventricular function is further compromised when the tricuspid valve annulus widens in the setting of RV dilatation, with failure of the tricuspid valve leaflets to coapt properly and worsening tricuspid regurgitation [24]. These changes result in a perpetuating cycle of increasing wall stress, worsening ischemia, tricuspid regurgitation, and unfavorable loading conditions that ultimately lead to RV failure [25] (Fig. 18.2).
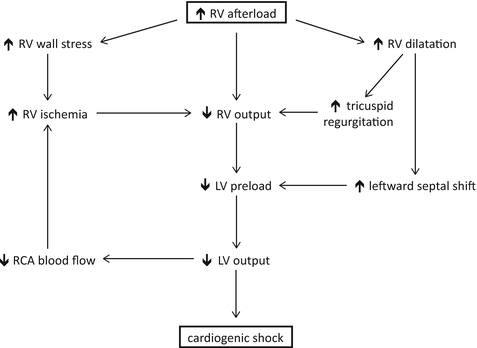
Fig. 18.2
Pathogenesis of right ventricular failure secondary to increased right ventricular afterload. RV right ventricle, LV left ventricle, RCA right coronary artery
Because the RV and LV share the space provided by the pericardium, increases in RV size are at the expense of LV volume. With RV dilatation, the interventricular septum undergoes leftward displacement, impinging upon the LV cavity. The septum’s encroachment on the LV impairs LV filling, and the distortion in LV geometry causes a decline in LV systolic performance [26–30]. In addition, the LV is only able to pump the stroke volume received from the right heart, which steadily declines in the failing RV. The resultant decrease in LV stroke volume and CO leads to a fall in RCA blood flow and an exacerbation of RV ischemia, fueling the RV failure cascade (Fig. 18.2).
Acute RV failure from rapid increases in RV afterload can occur in the setting of massive pulmonary embolism (PE). In patients without prior cardiopulmonary disease, good correlation has been observed between hemodynamics and the degree of angiographic obstruction from PE [31–33]. Mean pulmonary artery (PA) pressures of greater than 20 mmHg occur when there is greater than 25–30 % obstruction of the pulmonary vascular bed. When more than 50 % of the pulmonary vascular bed is occluded acutely, patients with are unable to generate mean PA pressures of greater than 40 mmHg, presumably the maximum pressure a normal RV can generate. Thus, when pulmonary vascular obstruction exceeds 50 %, the inability of the RV to generate higher pressures leads to a decrease in CO and the initiation of the RV failure cascade [31, 34]. Mean PA pressures of greater than 40 mmHg suggest that the RV has hypertrophied from chronic increases in afterload such as those caused by underlying cardiopulmonary disease [17]. Patients with poor cardiopulmonary status, manifest a greater deterioration in hemodynamics with a lesser degree of pulmonary vascular obstruction [35]. In patients with PAH, venous stasis, a sedentary lifestyle, dilated right heart chambers, and sluggish pulmonary blood flow together increase the risk for intrapulmonary thrombosis and thromboembolism.
RV failure can also occur in the absence of increased afterload, as seen in RV infarction. RV infarction results in both decreased RV systolic performance from profound depression of RV free wall contraction as well as RV diastolic dysfunction from ischemia [36, 37]. The culprit vessel for RV infarction is usually the RCA and significant RV infarction nearly always occurs in the setting of acute transmural inferior-posterior LV infarction [38]. When the RCA lesion is proximal to the right atrial (RA) branches, RA ischemia can impair RA function, further worsening right-sided hemodynamics [30, 36, 39]. With RV dilatation and decreased RV output, an RV failure cascade similar to that described above can occur. In the absence of preexisting pulmonary vascular disease or significant LV dysfunction, RV systolic and PA pressures do not rise. As LV septal contraction augments RV systolic pressure, concomitant LV infarction, particularly if the septum is involved, may result in further hemodynamic compromise [36, 40]. The short-term prognosis of patients with RV infarction is poor, owing to a higher incidence of cardiogenic shock, ventricular arrhythmias, and high-grade atrioventricular block. The increase in short-term mortality appears to be related to the actual presence of RV infarction and not total myocardial infarct size [41–46]. On the other hand, for patients who survive the acute period, the long-term prognosis is quite favorable even without successful coronary intervention, as RV function is able to return to near normal both at rest and with exercise [47, 48]. This dramatic improvement in RV function in the absence of reperfusion in survivors may be due to transient RV ischemia and myocardial stunning in the acute period, not true RV infarction [49–51]. The RV is less susceptible to true infarction for several reasons. First, its smaller mass and lower afterload result in lesser oxygen demand; Second, it receives coronary perfusion continuously throughout both diastole and systole; Finally, it has more extensive collateral flow from the left to the right coronary arteries. In fact, chronic RV failure attributable solely to RV infarction is a very rare occurrence [9, 51–53].
Triggers for RV Failure
A patient with preexisting pulmonary vascular and RV dysfunction can deteriorate rapidly when challenged by various insults. The prompt identification of contributing or inciting factors is therefore critical in the successful management of these patients [19]. A broad differential diagnosis should be considered because clinical worsening in these patients may be due to a variety of multisystem derangements and etiologies.
A rigorous search for potential sources of infection should be conducted when patients with PH become acutely ill. In a patient with chronic RV dysfunction and venous congestion, reduced bowel perfusion can impair the function of the intestinal barrier, leading to translocation of bacteria and/or release of endotoxin [54]. Patients with chronic PAH who have indwelling central venous catheters for the administration of vasoactive therapies are at increased risk for developing catheter-related skin and soft tissue infections as well as bacteremia [19]. Chronic PAH therapies, specifically prostacyclins, may also have immunosuppressive effects [55–58]. Pneumonia is particularly problematic in patients with pulmonary vascular disease because the resultant shunt with hypoxia can lead to vasoconstriction and further elevation in pulmonary vascular resistance (PVR).
The hemodynamic perturbations in sepsis can cause profound deterioration in patients with severe PH and RV failure. The increase in capillary leak and venous capacitance observed in sepsis results in decreased venous return and lower RV filling pressures, a condition poorly tolerated by a failing RV. The failing RV relies heavily on elevated end diastolic pressures to maintain cardiac output and is sensitive to abrupt changes in loading conditions [59]. Systemic hypotension from sepsis-induced peripheral vasodilation can result in decreased RCA blood flow, exacerbating RV ischemia and compromising RV function. Even in the absence of preexisting pulmonary vascular disease, sepsis has been shown to cause RV myocardial dysfunction directly through cytokine-mediated myocardial depression, as well as indirectly via hypoxic pulmonary vasoconstriction from associated lung injury [60–63]. As discussed later in the text, the addition of vasopressors and inotropes are fraught with their own difficulties in this hemodynamically fragile patient population. Irrespective of the site of infection, the development of sepsis can be devastating and must be managed aggressively, as described below. For these reasons, when infection is suspected, clinicians should have a low threshold to promptly administer antibiotics.
Acute respiratory distress syndrome (ARDS) can lead to the development of RV dysfunction, primarily via increases in RV afterload [64–70]. A variety of factors contribute to pulmonary vascular dysfunction in ARDS, namely, pulmonary vasoconstriction from hypercapnia and hypoxia, and increased intra-thoracic pressures secondary to mechanical ventilation and high positive end-expiratory pressures [70, 71]. Thrombosis also contributes to the increased RV afterload as postmortem studies have demonstrated thromboemboli in 95 % of patients with ARDS [65, 72]. A sub-study from a large clinical trial of ARDS found that 73 % of patients with ARDS had pulmonary vascular dysfunction, as evidenced by an increase in the transpulmonary gradient (TPG) and pulmonary vascular resistance index. The presence of pulmonary vascular dysfunction was independently associated with increased mortality in a dose-dependent fashion, such that increasing TPG was associated with higher mortality [64]. Although the mortality rates of patients with chronic PH who develop ARDS have not been well studied, it seems likely that PH patients may be particularly susceptible to poor outcomes in the setting of ARDS.
Atrial tachyarrhythmias may contribute to hemodynamic compromise in critically ill patients with pulmonary vascular disease. In a cohort of 231 patients with PAH or inoperable CTEPH, the annual incidence of new-onset supraventricular tachycardia was 2.8 %, most commonly due to atrial fibrillation or atrial flutter [73]. Because patients with PH may already have impaired LV filling, these tachyarrhythmias are poorly tolerated and can lead to clinical decompensation. In addition, the loss of atrial contractions may be deleterious in the setting of a noncompliant RV [25, 74]. The maintenance or restoration of sinus rhythm may be helpful in the management strategy discussed below, although data supporting this approach are lacking.
Other factors that may contribute to RV failure in the setting of PH include anemia, hypoxemia, hypercapnia, acidosis, PE, and metabolic abnormalities [24]. Iatrogenic causes include the abrupt withdrawal of pulmonary vasodilators leading to rebound PH, the administration of medications with negative inotropic properties such as β-blockers and certain calcium channel blockers, and the use of positive pressure ventilation, especially with high mean airway pressures and volumes [19]. In a study of 46 patients with PAH or inoperable CTEPH admitted to the ICU, 19 patients had an identifiable trigger for RV decompensation, of which 11 had infection, 3 had unplanned modification or withdrawal of pulmonary vasodilator therapy, 1 had unplanned withdrawal of diuretics, 3 had cardiac arrhythmias, and 1 had unplanned pregnancy [1].
Monitoring in the ICU
Close monitoring of patients with PH and RV failure is critical and may incorporate a mix of noninvasive and invasive modalities. The use of cardiac biomarkers may help in risk assessment because natriuretic peptides are secreted in the setting of right atrial and RV myocardial stretching and cardiac troponin is released during myocardial necrosis from RV ischemia [75]. Elevations in troponin and natriuretic peptides are associated with worse outcomes in chronic PH and acute pulmonary embolism [21, 75–82]. In a prospective cohort study examining 46 patients with PAH or inoperable CTEPH high brain natriuretic peptide levels on admission to the ICU were associated with increased mortality, although no statistical link was found between troponin levels and survival [1].
Echocardiography is a noninvasive modality that plays a central role in the management of patients with PH by providing information about RV function and geometry. In addition, it can help elucidate possible precipitating factors of RV failure, including LV dysfunction and valvular disease. Common echocardiographic signs of RV failure include RV dilatation with associated loss of its typical triangular shape and paradoxical motion of the interventricular septum during systole [83]. In chronic PAH, echocardiographic predictors of poor outcomes include RA enlargement, pericardial effusion, low tricuspid annular plane systolic excursion, and septal displacement, although their ability to discriminate between patients who do well and those who deteriorate in the acute care setting remains unclear [84–86].
The presence of a large pericardial effusion in critically ill patients with pulmonary vascular disease and/or RV failure can be alarming, particularly in the setting of decreased cardiac output or hypotension. The question of “silent tamponade” often arises because elevated RVEDP may prevent the normal echocardiographic findings of RV collapse during inspiration. However, drainage of pericardial effusions in PAH patients traditionally has not been advised due to high reported mortality rates following pericardiocentesis or the placement of a pericardial window [87]. Acute RV decompensation following removal of pericardial fluid likely occurs because of a sudden increase in RV transmural pressure. Under normal conditions the RV is well suited to accommodate this relative increase in RV filling pressure, but in the acutely or chronically overloaded RV, filling pressures have often reached extremely high levels and any further increase may result in a fall in RV contractility. In fact, pericardial effusion in chronic RV failure is often caused by high RVEDP that impedes drainage of pericardial fluid [87]. Gradual removal of fluid may obviate this concern. A more recent single center experience with pericardiocentesis in PAH found low procedural mortality, suggesting that in highly experienced hands there may be a role for this procedure if tamponade is suspected [88]. However, extreme caution is advised.
For patients with PH presenting in shock, it is imperative to obtain adequate central venous access and to perform frequent and blood pressure monitoring. Pulmonary artery catheterization (PAC) may help to differentia maldistributive from cardiogenic shock as well as guide ICU therapy. For example, the measurement of PA oxygen saturation, a marker of the adequacy of oxygen delivery for the body’s oxygen demand, may determine the need for inotropes [83].
The use of PAC in critically ill patients without PH has declined over time because multiple studies have shown that outcomes are not improved with its use [89–92]. However, no studies have been carried out in the acute setting for the “pulmonary vascular” population and obtaining and following pulmonary hemodynamics may be important during acute illness for certain PH patients [90]. As the RV is usually severely dilated in advanced PAH, it is advisable to use fluoroscopy to guide the PA catheter into place in order to avoid excessive ectopy and arrhythmias. It is important to note that the PA diastolic pressure cannot be substituted for the pulmonary capillary wedge pressure in patients with PAH due to the presence of a significant TPG. An accurate pulmonary capillary wedge pressure can be difficult to obtain in patients with PAH but is a critical value and requires due diligence, as the approach to management differs markedly in PAH as compared to pulmonary venous hypertension.
Management
General management goals for patients with PH and RV failure include optimization of RV preload, maintenance of adequate mean systemic arterial pressure, enhancement of RV contractility, and reduction of RV afterload, while treating any potentially reversible causes for the acute decompensation [90]. Unfortunately, PH in the ICU setting has not been robustly studied, and consensus guidelines are lacking. Management strategies therefore rely heavily on the guidance of experienced PH specialists.
Volume Management
Optimizing preload in patients with RV failure is complex because both hypovolemia and hypervolemia can have detrimental effects on CO. In most but not all cases of RV failure, the dilated and stretched RV is operating on the flat portion of the Frank-Starling curve. Volume expansion in this scenario often does not augment CO and can instead exacerbate RV dilatation, increase RV wall tension, worsen tricuspid regurgitation, and displace the inter-ventricular septum toward the LV. These adverse effects on RV function and LV filling contribute to reducing CO and worsening clinical status. The goal, therefore, should be to maintain a negative fluid balance with the use of diuretics and, if necessary, hemofiltration, to decrease the volume load on the distended and failing RV without compromising preload. Adjustments in the rate and amount of volume removal should be made according to hemodynamic response [12, 93–97]. In the case of clear intravascular volume depletion, a conservative strategy of holding diuretics, encouraging oral hydration (if applicable), or judiciously using small fluid boluses is preferred.
Vasopressors
Mean systemic arterial pressure must be maintained to minimize RV ischemia. Increased RV wall stress leads to RCA hypoperfusion as PVR approaches SVR or as SVR falls in mixed shock states (i.e., vasodilatory shock with concomitant RV failure). If PVR exceeds SVR, RCA perfusion will occur only during diastole, exacerbating RV ischemia. With the use of vasopressors, SVR can be increased, leading to augmentation of mean systemic arterial pressure, lowering of the PVR/SVR ratio, and, ultimately, improvement in RV myocardial perfusion [9, 90, 98]. In addition, the use of vasopressors increases LV afterload, helping to normalize distorted LV geometry from a leftward-bowed septum [25]. In choosing a vasopressor agent, it is important to be aware of each drug’s effect on PVR because increases in PVR may contribute to clinical decompensation.
Norepinephrine causes vasoconstriction through the α1 receptor and has limited inotropic properties from β1 receptor stimulation [99]. It has been shown to improve RV performance and PA/RV coupling in animal models of acute PH and RV dysfunction by way of β1 effects on contractility [20, 100, 101]. In a study of ten patients with septic shock, PH, and RV dysfunction, norepinephrine increased SVR and improved the RV oxygen supply/demand ratio but also caused an increase in PVR and failed to improve RV ejection fraction [102]. Evidence for the use of norepinephrine in critically ill patients with PH comes from a large randomized trial of ICU patients with shock, in which norepinephrine use, as compared to dopamine use, was associated with decreased mortality at 28 days in the prespecified subgroup of patients with cardiogenic shock and with a decreased rate of arrhythmias in all patients [103].
Phenylephrine, an α1 receptor agonist with no β1 receptor activity, improves RCA perfusion in RV failure, but this benefit is offset by its elevation of PVR and lack of β1-mediated enhancement of contractility [98, 100, 104]. In addition, reflex bradycardia secondary to phenylephrine may have detrimental effects in the setting of RV failure [105].
Epinephrine, a potent α and β receptor agonist that causes vasoconstriction and increased inotropy, increased CO without altering the PVR/SVR ratio in hypoxic newborn piglets [106]. A small study in patients with RV dysfunction from severe septic shock showed that epinephrine increased RV contractility [107]. The use of epinephrine has not been well studied in patients with PH, however. For these reasons, norepinephrine is a more favorable choice for patients with PH in the ICU than either phenylephrine or epinephrine.
Vasopressin causes vasoconstriction by acting upon V1 receptors on vascular smooth muscle cells and also increases vascular responsiveness to catecholamines [99]. Low doses of vasopressin cause pulmonary vasodilation through endothelium-mediated nitric oxide (NO) production in animals, although high doses cause vasoconstriction through an endothelium-independent mechanism [108]. Vasopressin’s effect on the pulmonary vasculature has been inconsistent in human studies [105]. At higher infusion rates, vasopressin may have direct myocardial depressive effects and causes coronary vasoconstriction [109, 110]. Although low-dose (i.e., 0.01–0.03 U min−1) vasopressin may be effective in managing patients with RV failure, higher doses should be used with caution.
Inotropes
Dopamine activates dopaminergic receptors at doses less than 5 μg kg−1 min−1, β1 receptors at doses between 5 and 10 μg kg−1 min−1, and α1 receptors at doses greater than 10 μg kg−1 min−1, although actual plasma dopamine levels for a given infusion rate may vary unpredictably in critically ill patients [99, 111]. In a large animal model of hypoxia-induced PH, dopamine did not increase PVR at doses up to 10 μg kg−1 min−1 [112]. In fact, in patients with PH secondary to left heart disease, dopamine in doses ranging from 2 to 16 μg kg−1 min−1 increased CO and heart rate but did not significantly change PVR [113]. Similarly, in a small study of patients with PH and septic shock, dopamine improved CO without increasing PVR but failed to improve RV ejection fraction [102]. As discussed above, although the routine use of dopamine in the critical care setting is not supported by current data, in low doses it may be a reasonable option in patients with PH and RV failure. Aggravation of tachycardia can be a limitation.
Dobutamine has inotropic effects through β1 receptor stimulation and a variable degree of vasodilatory effects through the β2 receptor [99]. In an animal model of PH, dobutamine at doses of 5–10 μg kg−1 min−1 restored CO and arterial pressure without affecting PA resistance or elastance, improving PA/RV coupling [20]. Doses up to 10 μg kg−1 min−1 in patients with left heart failure result in improved myocardial contractility, reduced PVR and SVR, and less tachycardia when compared with dopamine [114]. Dobutamine has been shown to improve hemodynamics in patients with PH at liver transplantation and after severe RV infarction [97, 115]. However, it has significant β2-mediated systemic vasodilatory properties and thus, it is important to avoid high doses, anticipate possible systemic hypotension, and be prepared to add systemic vasopressors if systemic hypotension occurs [90].
Milrinone, a selective phosphodiesterase-3 inhibitor, that acts via delaying metabolism of intracellular cAMP has positive inotropic effects and direct-acting vasodilatory properties on the pulmonary circulation [99]. In animal models of both acute and chronic PH, milrinone improved RV function and decreased PVR [116, 117]. In patients with pulmonary vascular dysfunction in the setting of LV failure, post-ventricular assist, or cardiac transplantation, milrinone has been shown to reduce pulmonary pressures and improve RV function and is often the agent of choice in these settings [118–120]. Systemic hypotension often limits the use of milrinone in the treatment of patients with PAH and hemodynamic instability, but it may be effective in patients with PH associated with LV dysfunction. Dopamine, dobutamine, and milrinone therapies are all capable of inducing cardiac tachyarrhythmias that are poorly tolerated in patients with PH and may be a limiting factor in their use in some patients. A few case series suggest that inhaled milrinone may be useful in PH because it minimizes systemic hypotension by delivering the drug directly to the pulmonary vasculature [121–124].
Levosimendan, a novel drug that enhances myocardial contractility by sensitizing troponin C to calcium while also acting as a pulmonary vasodilator, is a promising agent for patients with PH and RV failure but has not yet been thoroughly investigated in this patient population [125–129]. Irrespective of the specific agent, inotropes should generally be considered when there is evidence of inadequate oxygen delivery and/or in the case of volume overload not successfully managed with diuretics alone. It is especially important to avoid “supra-normalization” of oxygen delivery in these patients because this strategy not only is associated with worse outcomes in the general ICU population but may also increase PA pressures and worsen cardiac function in patients with pulmonary vascular disease [130, 131].
Pulmonary Vasodilators
Because increased RV afterload plays a central role in RV failure associated with PH, the use of pulmonary vasodilators to unload the RV is critical. The ability of even a severely dilated and overloaded RV to return to normal size and function is illustrated by the restoration of RV function after pulmonary thromboendarterectomy for CTEPH and lung transplantation in patients with PH [132–134].
Inhaled NO is a potent pulmonary vasodilator with minimal systemic vasodilatory effects because it is rapidly inactivated by hemoglobin within the pulmonary capillaries. Because of its short half-life, continuous administration through a face mask, a nasal cannula, or, a ventilator circuit is required [135]. In patients with chronic PAH, NO reduces PVR and improves CO without a drop in SVR [136–138]. In 26 patients admitted to the ICU with acute RV failure, NO administration resulted in a greater than 20 % increase in CO and/or decrease in PVR in half of the patients [139]. With prolonged use at high concentrations, methemoglobinemia may develop, necessitating periodic surveillance of methemoglobin levels and routine assessment for cyanosis [140]. Nitrogen dioxide (NO2) will accumulate when NO is delivered with high FiO2 and needs to be monitored continuously in the ventilator circuit. Care must be taken in the discontinuation of NO because abrupt withdrawal has been associated with rebound PH and hemodynamic collapse [141–143]. After prolonged use, complete discontinuation of even low-dose inhaled NO may necessitate bridging therapy with another targeted pulmonary vasodilator.
Prostacyclins, including epoprostenol, treprostinil, and iloprost, are potent, short-acting agents that cause pulmonary vasodilation and inhibit platelet aggregation. In chronic PAH, these medications improve exercise capacity, hemodynamics, and, in the case of continuous infusion epoprostenol, survival [144–149]. In the critical care setting, prostacyclins have been mainly studied in patients with PH after cardiac surgery or transplantation, where they have been shown to reduce PVR and improve RV function [150–156]. The use of intravenous prostacyclins and up-titration of their dose in the ICU is usually limited by systemic hypotension and other systemic adverse effects including nausea, flushing, headache, and diarrhea [55]. Prostacyclins should be avoided in patients with significant left heart dysfunction and elevated pulmonary venous pressures because their use in this setting can generate further increases in left-sided filling pressures, leading to the development of pulmonary edema, pleural effusions and/or the deterioration of LV function [157].
Importantly, vasodilation of the pulmonary vasculature by systemic prostacyclin or prostacyclin analogues is nonselective and can exacerbate ventilation-perfusion mismatch, leading to worsened gas exchange and hypoxemia. These effects may be particularly problematic in patients with intrinsic lung disease and hypoxic PH. This phenomenon maybe circumvented with the use of inhaled preparations whereby pulmonary blood flow to well-ventilated regions in the lung is increased, thereby decreasing intra pulmonary shunt [158]. The inhaled prostacyclins iloprost and treprostinil are approved for outpatient use with specific devices (the I-neb Adaptive Aerosol Delivery device and the Optineb-ir, respectively). Iloprost is increasingly being used “off-label” in the postoperative and ICU setting with an ultrasonic nebulizer, although dosing and drug absorption are not standardized. Drug delivery and pharmacokinetics using alternative systems (i.e., conventional nebulizers and ventilator circuits) have also not been studied. Finally, abrupt discontinuation of prostacyclin infusions in chronically treated patients should be avoided because this may precipitate severe rebound PAH and even death [159]. Patients with PAH on chronic prostacyclin therapies who develop vasodilatory or mixed shock may require dose reductions during their acute illness, so as to not exacerbate systemic hypotension and/or create (relative) high-output failure, but this should be done under the guidance of a PH specialist.
The endothelin receptor antagonists and phosphodiesterase-5 (PDE5) inhibitors are proven oral medications for the management of chronic PAH, but they have not been investigated in critically ill patients with RV failure [160–163]. An intravenous formulation of the PDE5 inhibitor sildenafil has recently become available and may prove useful in the acute setting, although future studies in this patient population are needed [164]. By increasing downstream cyclic guanosine monophosphate signaling, PDE5 inhibitors reduce PVR and may also augment RV function by exerting a milrinone-like effect through inhibition of phosphodiesterase-3 [165–168]. With a rapid onset of action and a relatively prolonged half-life of 3–4 h, intravenous sildenafil must be used cautiously in critically ill patients to avoid systemic hypotension and exacerbation of ventilation-perfusion mismatch [164, 169]. Anecdotally, intravenous dosing should be approximately one third to one half that of the anticipated oral sildenafil dose.
Supportive Care
Maintaining peripheral oxygen saturations greater than 90 % in PAH; this is critically important in the acute setting, to prevent or reverse any contributing hypoxic pulmonary vasoconstriction [170]. Oxygen inhalation has been shown to reduce PVR and improve CO in patients with PH [171, 172]. Hypercapnia and acidemia worsen hypoxic pulmonary vasoconstriction and RV contractility and should be avoided [173, 174]. Although no studies have been performed to determine the optimal hemoglobin level for patients with PH and RV failure, many experts recommend maintaining a hemoglobin level of greater than 10 g dL−1 to optimize oxygen-carrying capacity and minimize RV ischemia [25]. Volume status should be monitored carefully and diuretics adjusted appropriately with transfusion. As mentioned previously, patients with PH and RV failure are intolerant of electrolyte disturbances, metabolic derangements, and vagal stimuli, and care must be taken to normalize these imbalances if present.
Mechanical Ventilation
Endotracheal intubation of patients with PH and RV failure should be avoided when at all possible. If intubation is necessary, etomidate is generally the preferred induction sedative because it has minimal effects on SVR, PVR, and cardiac contractility. Propofol should be avoided given its propensity to cause systemic hypotension. Systemic vasopressors should be readily available or started preemptively to maintain SVR and counteract peri-intubation hypotension, which, if occurs in a patient with severe PH and RV failure, can be catastrophic and result in cardiac arrest [175, 176].
Because elevated intrathoracic pressures decrease RV and LV preload and increase PVR, ventilator strategies that use high inflation lung volumes and pressures should be avoided to prevent these detrimental effects [90, 177]. High positive end-expiratory pressures may, in theory, increase pulmonary vascular resistance and should be avoided, as should atelectasis or reduced lung volumes. In general, ventilation strategies should aim to keep lung volumes close to Functional Residual Capacity (FRC) while maintaining adequate oxygenation and ventilation while avoiding high peak inspiratory or end expiratory pressures.
Mechanical Support
When conventional support for the RV is ineffective in severe cases of PH and RV failure, mechanical support, including RV-assist devices and venoarterial extracorporeal membrane oxygenation (VA-ECMO), may be considered. The use of intra-aortic balloon counterpulsation in isolated RV failure has been reported, and improves CO by augmenting coronary blood flow [178, 179]. Although RV-assist devices are effective in primary RV dysfunction or RV failure secondary to LV failure, thus far, they have not been shown to be successful in patients with significantly elevated PVR. In this setting, the increase in pulmonary blood flow, particularly if pulsatile, gives rise to markedly elevated pulmonary vascular pressures, which can damage the pulmonary microcirculation and lead to intraparenchymal pulmonary hemorrhage, hemoptysis, and death [180, 181].
VA-ECMO has been used successfully in patients with massive PE, in patients with PH and RV failure as a bridge to lung transplantation, and in patients with CTEPH as a bridge to or for complications arising after pulmonary thromboendarterectomy [182–189]. By unloading the RV and providing additional blood flow to the systemic circulation, VA-ECMO improves the hemodynamics of patients with PH and RV failure. In addition, it can help reverse hypoxic pulmonary vasoconstriction by performing gas exchange and has been successfully used in awake, spontaneously breathing patients (a number of whom had PAH) as a bridge to lung transplantation [190]. VA-ECMO is not without potentially serious complications, however, and may include bleeding, infection, thromboembolism, and neurologic sequelae [180]. More recently, pumpless lung-assist devices have been developed and used in patients with PAH as a bridge to lung transplantation, connecting the PA to the left atrium with a low-resistance membrane oxygenator. With the blood flow through the circuit powered by the patient’s own RV, these devices unload the RV and enhance LV filling in a manner similar to balloon atrioseptostomy; however, in contrast to septostomy, these devices improve instead of impair gas exchange [191, 192].
Cardiopulmonary Resuscitation
Cardiopulmonary resuscitation (CPR) in patients with PH and RV failure is largely unsuccessful. In a retrospective, multicenter study, 132 patients with PAH had circulatory arrest and implementation of CPR, of which only 8 patients (6 %) survived for more than 90 days. In addition, seven of these eight surviving patients had correctable causes of their cardiopulmonary arrest, including vasovagal reactions, digitalis toxicity, and pericardial tamponade [193]. One explanation for the lack of efficacy of CPR in this patient population is that high PVR makes it unlikely that chest compressions will achieve adequate pulmonary blood flow and LV filling. In addition, the use of epinephrine during CPR causes even further increases in PVR [194]. In light of the poor outcomes of CPR in patients with severe PH and RV failure, having timely discussions about the possibility of “Do Not Resuscitate” orders with these patients and their families is crucial, particularly in patients in whom a reversible cause for decompensation cannot be determined.
Conclusion
Patients with PH and RV failure who require ICU admission are at significant risk for worsening morbidity and mortality. In the setting of PH, RV failure can be triggered by various factors, giving rise to a vicious cycle of worsening RV function, profound shock, and death. Given their tenuous hemodynamics, these patients must be monitored closely with indicators of end-organ perfusion, cardiac biomarkers, echocardiography, and, in select situations, PAC. Management goals include optimizing RV preload with diuretics or hemofiltration, maintaining mean systemic arterial pressure with systemic vasopressors, augmenting RV contractility with inotropes, decreasing RV afterload with selective pulmonary vasodilators, and reversing any identifiable inciting factors. In severe cases, VA-ECMO or other forms of mechanical support may be considered. Although there have been marked advances in therapeutic strategies for PAH over the last several decades, acute care of patients with pulmonary vascular and RV dysfunction remains largely unstudied and guided by clinical expertise.
References
1.
Sztrymf B, et al. Prognostic factors of acute heart failure in patients with pulmonary arterial hypertension. Eur Respir J. 2010;35(6):1286–93.PubMed
2.
Kurzyna M, et al. Characteristics and prognosis of patients with decompensated right ventricular failure during the course of pulmonary hypertension. Kardiol Pol. 2008;66(10):1033–9. discussion 1040–1.PubMed
3.
Huynh TN, et al. Prognostic factors and outcomes of patients with pulmonary hypertension admitted to the intensive care unit. J Crit Care. 2012;27(6):739e7–13.
4.
Greyson CR. Pathophysiology of right ventricular failure. Crit Care Med. 2008;36(1 Suppl):S57–65.PubMed
5.
Zaffran S, et al. Right ventricular myocardium derives from the anterior heart field. Circ Res. 2004;95(3):261–8.PubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


