Fig. 8.1
Metabolic pathways reprogrammed in PAH. Several pathways are affected in humans with and experimental models of PAH. Increased glycolysis, lactate fermentation, and glutaminolysis are observed (purple, pink, and blue), while fatty acid oxidation (FAO) is decreased (green). Glycolysis can decrease FAO through increases in cytoplasmic malonyl-CoA inhibiting carnitine palmitoyl transferase 1 (CPT1) (orange). Levels of cytoplasmic malonyl-CoA are carefully regulated by the activities of acetyl-CoA carboxylase (ACC) and malonyl-CoA decarboxylase (MCD). FAO inhibits glycolysis as a consequence of citrate accumulation. Dichloroacetate (DCA) shuttles glycolytic carbon into the TCA cycle (red) by inhibiting pyruvate dehydrogenase kinase (PDK), an inhibitor of pyruvate dehydrogenase (PDH). The 3-ketoacyl CoA thiolase inhibitors (KATi), decrease FAO and relieve its inhibition of glycolysis to facilitate glucose oxidation. A glutamine analogue, 6-diazo-5-oxo-l-norleucine (DON) has been used in experimental models to improve RV function by inhibiting glutaminolysis
With the dramatic resurgence of investigations that incorporate molecular metabolism as key pieces of both theoretical and experimental design, it has become common practice to use “glycolysis” as shorthand for what is perhaps more properly described as “lactatogenic glycolysis” – glycolysis that ultimately results in the generation of lactate from the reduction of pyruvate by LDH. The terms “anaerobic glycolysis” and “aerobic glycolysis” are not especially helpful, as glycolysis is occurring regardless of oxygen availability. The intended distinction arises from the fate of pyruvate, whether it is oxidized in the TCA cycle (glucose oxidation, an aerobic process) or fermented to lactate by LDH (lactate fermentation, an anaerobic process) (Fig. 8.1). For the purposes of this discussion, the term “glycolysis” will be used in the strict sense to refer to the biochemical process of converting glucose to pyruvate.
In the early twentieth century, Otto Warburg described the phenomenon by which cells that have undergone malignant transformation change their metabolic profile to utilize lactatogenic glycolysis as the primary mode of generating ATP [1, 2]. This is a normal mode of metabolism for mammalian cells under anaerobic conditions, when oxygen deficiency is limiting oxidative metabolism of carbon subunits. However, Warburg observed this type of anaerobic metabolism in malignant transformed cells growing in oxygen concentrations that were more than sufficient to permit oxidative metabolism. He concluded that the metabolic shift from glucose oxidation to lactatogenic glycolysis for ATP production – a phenomenon commonly referred to as the Warburg effect – is a primary driver of and a required adaptation for malignant transformation.
More recently, hypoxia-inducible factor 1α (HIF) has been identified as the principal regulator of the Warburg effect in a variety of cancer cells [3]. In normal cells, as oxygen tension falls, the HIF transcription factor is stabilized, thereby activating a gene expression program leading to (1) increased glucose uptake through the expression of glucose transporters; (2) increased glycolytic flux through the expression of several glycolytic enzymes (e.g., aldolase A, phosphoglycerate kinase 1, enolase 1, triosephosphate isomerase), including the key regulators of glycolytic flux, hexokinase, phosphofructokinase L, and pyruvate kinase M [4–8]; (3) decreased oxidative phosphorylation through expression of pyruvate dehydrogenase kinases (PDK) 1–4, inhibitors of PDH [9–11]; (4) increased lactate fermentation through expression of LDHA; and (5) increased mitochondrial autophagy through expression of BNIP3 [12]. The overall goal of this gene expression program is to mitigate reactive oxygen species (ROS) generation from dysfunctional mitochondrial electron transport chains [13]. Increasing evidence suggests that abnormal HIF stabilization under normoxic conditions is responsible for the Warburg effect in malignant cells [5, 7, 9, 10, 14].
Precisely this same type of metabolic reprogramming has been observed in PAH in both the pulmonary vasculature and the RV. The earliest evidence for a shift to lactatogenic glycolysis in experimental PAH came, perhaps not surprisingly, from the chronic hypoxia rat model. With chronic hypoxia, the RV showed increased expression and activity of hexokinase, the first and rate-limiting enzyme in glycolysis, as well as a modest increase in the expression of LDH [15]. Additional evidence of increased lactatogenic glycolysis was provided by the monocrotaline (MCT) rat model of PAH, where MCT treatment increased glycolysis and decreased FAO in cultured cardiomyocytes in a HIF-dependent manner [16]. HIF was already known to regulate glycolysis and to play some role in the pathogenesis of PAH [17–20], but this was one of the first demonstrations of a pathogenic link between glycolysis and HIF in a model of PAH.
Soon after, the observation of increased glycolysis in PAH was extended to include several different animal models of PAH as well as patients with the disease. In the Fawn hooded rat model (FHR), dysmorphic and hyperpolarized mitochondria with deficient superoxide production drove normoxic activation of HIF in pulmonary artery smooth muscle cells (PASMCs) [21]. Normoxic HIF activation was confirmed in PASMCs cultured from PAH patients. One of the implications of this study was that a glycolytic shift mediated by HIF occurs in PASMCs in PAH, but this was not directly demonstrated.
Subsequently, increased glycolytic carbon flow and lactate production were demonstrated in pulmonary artery endothelial cells (PAECs) cultured from PAH patients [22]. Positron emission tomographic imaging (PET) showed that uptake of 18 F-fluorodeoxyglucose (FDG) in the lungs of PAH patients was significantly increased compared to healthy controls, providing at least indirect evidence that the glycolytic reprogramming observed in vitro was also occurring in vivo in humans. As with cultured cardiomyocytes and PASMCs, the glycolytic shift in PAECs is dependent upon normoxic activation of HIF. The finding of increased glucose uptake in the lungs measured by FDG-PET has since been replicated in experimental PAH [23].
The mechanisms of normoxic HIF activation remain unclear. One hypothesis is that that epigenetic silencing of superoxide dismutase 2 (SOD2), the mitochondrial enzyme responsible for H2O2 generation from superoxide, leads to HIF activation through decreased cellular H2O2 production (or increased superoxide levels) (see below, Fig. 8.2) [24].
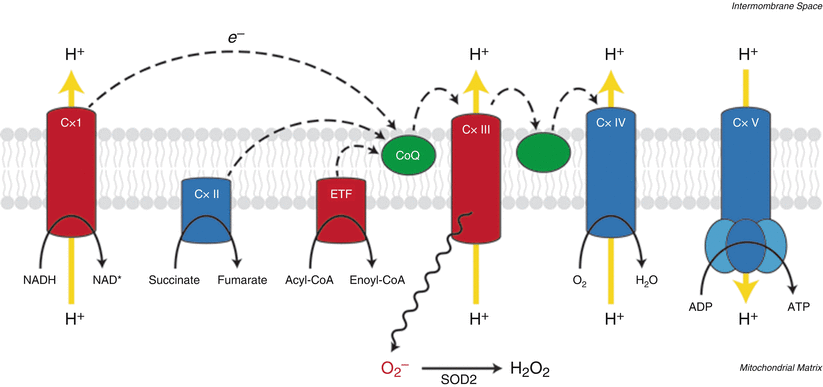
Fig. 8.2
Electron transport chain and oxidative phosphorylation. The electron transport chain transfers electrons (e −, dashed lines) from high-energy reduced carriers to molecular oxygen coupled to the generation of a proton gradient (yellow arrows) across the inner mitochondrial membrane. This proton gradient drives phosphorylation of ADP by ATP synthase (Cx V). Superoxide anion (O2 −) can be generated by complexes I and III and by electron transferring flavoprotein dehydrogenase (ETF) (red symbols; shown only for Cx III for clarity). Dismutation of superoxide is catalyzed by superoxide dismutase 2 (SOD2) to form hydrogen peroxide. Epigenetic silencing of SOD2, and reduced H2O2 formation, has been implicated in HIF activation in the pulmonary vasculature of patients with PAH
Similar to the pulmonary vasculature, evidence suggests that the pressure-overloaded RV has increased glycolytic flux. The MCT and pulmonary artery banding (PAB) rat models of pulmonary hypertension demonstrated increased FDG uptake and increased Glut1 expression [23, 25]. However, FDG-PET studies of the RV in patients with pulmonary hypertension have shown conflicting results [26]. Some studies have indicated that FDG uptake in the RV is increased compared to the LV in patients with pulmonary hypertension and that the uptake is proportional to disease severity [27–29], while others have not replicated these findings [30].
In contrast to the pulmonary vasculature, the role of HIF activation in mediating this metabolic change in the RV is controversial, as HIF expression has not been consistently shown to be elevated in animal models of pulmonary hypertension. No change in HIF was observed in the RV of the FHR [31], while increased HIF was observed in the compensated hypertrophic RV of the MCT rat prior to RV decompensation, whereafter HIF expression was noted to decrease [25]. PDK4 is the isoform primarily responsible for the metabolic switch from glucose oxidation to lactate fermentation in the RV and it is not known to be a direct target of HIF (although PDK4 expression does increase in hypoxia in some settings [11, 32]) [23, 31]. In the FHR model, PDK4 expression is likely driven by increased FOXO1, transcription factor expression and activity, rather than HIF [31].
Tricarboxylic Acid Cycle, Glutaminolysis, and Amino Acid Metabolism
The tricarboxylic acid (TCA) cycle, also known as Krebs’s cycle or the citric acid cycle, was one of the first cyclical metabolic pathways identified [33] (Fig. 8.1). The cycle is a central point of integration and regulation of carbon flow through multiple pathways within the cellular metabolic network. Regulation of carbon flow into and out of the TCA cycle is intimately linked to the balance of energy production, synthesis of biological macromolecules, cell proliferation, apoptosis, and redox homeostasis.
Classical teaching is that pyruvate generated from glycolysis is converted to acetyl-CoA by PDH, and this is the entry point into the TCA cycle. Acetyl-CoA condenses with oxaloacetate to form citrate (regenerating citrate, thus, the “citric acid cycle”), which is then converted through a series of sequential enzymatic reactions back to oxaloacetate. For each turn of the cycle, three molecules of NAD+ are reduced to NADH, one molecule of GTP is produced from GDP and phosphate, one molecule of ubiquinone (oxidized coenzyme Q) is reduced to ubiquinol (reduced coenzyme Q), and two molecules of CO2 are liberated.
However, carbon flow both into and out of the TCA cycle is much more complex and dynamic than simple pyruvate entry via acetyl-CoA. The breakdown of amino acids and fatty acids can also contribute carbon to the TCA cycle. In general, reactions or processes that result in a net flow of carbon into the TCA cycle are termed anaplerotic reactions or anaplerosis. (Note that some sources will use this term more specifically to refer to reactions that allow carbon entry into the TCA cycle without going through acetyl-CoA as an intermediate.) The major routes for anaplerosis include (1) pyruvate directly to oxaloacetate by pyruvate carboxylase, (2) l-glutamate to 2-oxoglutarate (2OG) by glutamate dehydrogenase, (3) β-oxidation of odd-chain fatty acids to succinyl-CoA as substrate for succinyl-CoA synthetase, (4) conversion of aspartate to oxaloacetate by aspartate aminotransferase, and (5) conversion of adenylosuccinate (an intermediate in purine nucleotide metabolism) to fumarate via adenylosuccinate lyase.
Reactions or processes that remove carbon from the TCA cycle to supply biosynthetic processes are termed cataplerotic reactions or cataplerosis. Carbons can exit the TCA cycle at virtually every point in the cycle to supply biosynthetic and regulatory reactions. For example, oxaloacetate and 2OG both support the synthesis of multiple amino acids. Citrate can be diverted out of the TCA cycle for the synthesis of fatty acids via ATP citrate lyase. Succinyl-CoA can be diverted from the TCA cycle to serve as the substrate for the enzymatic succinylation of lysine residues in proteins, a very recently recognized mechanism for post-translational regulation of protein structure and function [34]. Fumarate has also been shown to participate in the non-enzymatic succinylation of cysteine residues in proteins [35–38]. Clearly then, regulation of the anaplerotic/cataplerotic balance in the TCA cycle is complex, and any perturbation of this balance can have a significant impact on multiple biochemical pathways.
Glutaminolysis is one form of anaplerosis that has been linked to animal models of PAH. Glutamine is an important source of cellular energy, carbon, and nitrogen, particularly in rapidly proliferating cells [39]. Intracellular glutamine is hydrolyzed to glutamate, which is either reduced or transaminated to form 2OG (Fig. 8.1). The 2OG can be transported into the mitochondrion to enter the TCA cycle for ATP generation or reduced and carboxylated to form isocitrate for fatty acid synthesis. In the setting of impaired PDH, the increase in malate from the glutamine-fed TCA cycle is oxidized to pyruvate by malic enzyme (ME). In comparison to the PAB model, MCT rats had increased expression of glutamine transporters (SLC1A5 and SLC7A5) and increased malic enzyme induced by activation of c-Myc as a result of RV ischemia [40]. This is similar to glutaminolysis in malignancy where c-Myc expression is also increased [41]. Glutamine transporters may also be increased in the RV of patients with PAH [40].
A broader perturbation of amino acid metabolism, including increased glutamine metabolism and changes in branched chain amino acid utilization, has been shown in human PAEC expressing mutant BMPR2 in vitro as well as in in vivo measurements of transpulmonary amino acid gradients at the time of diagnostic right heart catheterization [42–44]. In contrast to c-Myc activation in the RV, normoxic activation of HIF in the pulmonary endothelium seems to be one of the key drivers of altered amino acid metabolism and altered TCA cycle carbon flux.
In addition to changes in the anaplerosis/cataplerosis balance, there is an emerging recognition of the role of novel metabolites as mediators of disease. This first came to prominence in the oncometabolism literature, with the observation that 2OG can be reduced to d-2-hydroxyglutaric acid (D2HG) in several malignancies as a result of a point mutation in the TCA cycle enzyme isocitrate dehydrogenase [45]. These mutations support oncogenesis through the inhibition of a variety of 2OG-dependent dioxygenase enzymes, such as HIF prolyl hydroxylase, histone demethylases, and TET-family 5-methylcytosine hydroxylases with a broad impact on epigenetic regulation of gene expression, collagen synthesis, and cell signaling [46, 47]. Very recently, l-2-hydroxyglutarate (L2HG) overproduction via reductive metabolism of 2OG has been demonstrated in response to hypoxia in PAECs and PASMCs [48]. This metabolite appears to have an important role in maintaining cellular redox homeostasis. In addition to the 2HG enantiomers, two other TCA metabolites, succinate and fumarate, also inhibit 2OG-dependent dioxygenase enzymes and may play an important role in tumorigenesis through their effect on cellular redox state [49]. Indeed, increases in succinate and other TCA metabolites have recently been shown in the lungs of PAH patients [50].
Fatty Acid Oxidation
Fatty acid oxidation is the primary metabolic fuel for the RV, and is another of the metabolic pathways affected by PAH (Fig. 8.1). In the low oxygen environment of the developing fetus, the heart relies on carbohydrate substrates that generate ATP through glucose and lactate oxidation [51]. However, shortly after birth, the heart shifts to FAO as the primary source of energy production. Fatty acids provide ~70 % of the energy for the resting heart with the balance generated from carbohydrate oxidation [52]. Importantly, the heart is flexible with regards to its energy supply; substrate availability, hormone levels (e.g., insulin, epinephrine), oxygen tension, and workload regulate substrate utilization. For example, increases in serum lactate during exercise are associated with increased lactate oxidation by the heart [53]. The balance between fatty acid and glucose consumption by the heart is carefully regulated by biochemical interactions between these two metabolic pathways in the so-called “glucose-fatty acid cycle” (or “Randle cycle”) initially proposed by Philip Randle and colleagues in 1963 [54, 55].
In isolated heart preparations, Randle and colleagues observed that utilization of fatty acids inhibits glucose oxidation and vice versa, all in the absence of hormonal regulation. Fatty acid oxidation increases mitochondrial ratios of acetyl-CoA/CoA and NADH/NAD+, which inhibit PDH (Fig. 8.1). Citrate accumulates in the cytosol, inhibiting phosphofructokinase, and the resulting accumulation of glucose-6-phosphate inhibits hexokinase. Fatty acid oxidation also inhibits glucose uptake by preventing translocation of glucose transporter 4 (GLUT4) to the plasma membrane; however, the mechanism for this effect remains undetermined [55].
Inhibition of FAO by glucose oxidation in the heart is mediated through increases in cytosolic malonyl-CoA. Malonyl-CoA inhibits carnitine palmitoyltransferase 1 (CPT1), which regulates the entry and oxidation of fatty acids in the mitochondria (Fig. 8.1). Cytosolic malonyl-CoA is determined by the complex regulation of acetyl-CoA carboxylase and malonyl-CoA decarboxylase (MCD), its synthetic and degradative enzymes, respectively. Notably, FAO inhibition by glucose oxidation in the heart requires an increase in glucose uptake through GLUT4 translocation to the plasma membrane as GLUT4 transport is saturated at circulating serum glucose concentrations [55].
In addition to suppressed glucose oxidation, FHRs have suppressed FAO as demonstrated by metabolic flux analysis in a working heart model and in freshly isolated cardiac myocytes [31], although PAB animals have increased FAO assessed by similar techniques [56]. A comparison of the VEGF receptor antagonist, Sugen 5416, plus hypoxia (SuH) and PAB rats demonstrated decreased expression of PGC-1α in SuH animals that was associated with decreased expression of acyl-CoA dehydrogenases, critical enzymes for fatty acid β-oxidation, while PAB animals had increased levels of these proteins [57]. FHRs may share similar inhibition of PGC-1α-regulated metabolic pathways. The transcription of FAO metabolic enzymes is also decreased in the MCT model [58]. Impaired fatty acid uptake has been observed in pulmonary hypertension patients using single photon emission computed tomography (SPECT) of a radiolabeled fatty acid analogue [59, 60]. There is also histologic evidence of impaired fatty acid utilization in the RV of PAH patients, as evidenced by abnormal lipid accumulation and lipotoxicity within the RV cardiac myocytes of PAH patients that is not present in RV samples from patients with left ventricular failure [61].
FAO may also play an important role in the pathogenesis of PAH in the vasculature. Mice deficient for malonyl-CoA decarboxylase (MCD), the main enzyme responsible for the degradation of malonyl-CoA, are protected from hypoxic pulmonary hypertension [25]. Decreased MCD activity increases cytosolic malonyl-CoA and inhibition of CPT1, thereby decreasing FAO and increasing glucose oxidation. Interestingly, a combined genetic and metabolomic analysis of human PAH lung tissue suggested increases in FAO due to increased expression of acyl-CoA dehydrogenases and FAO intermediates [62]. Additional studies are required to characterize the role of FAO in the pathogenesis of PAH in these two tissues, RV and pulmonary vasculature, particularly in consideration of potential therapies targeted at these pathways.
Oxidative Phosphorylation, Reactive Oxygen Species, and Mitochondrial Dynamics
The net consequence of the metabolic pathways discussed above is to allow electrons to enter the electron transport system within the mitochondria (Fig. 8.2). Here, electrons are transferred from high-energy reduced carriers (such as NADH synthesized in the TCA cycle) to ubiquinone (oxidized coenzyme Q). In the process, coenzyme Q is reduced to ubiquinol, and the carriers become oxidized (e.g., NADH is oxidized to NAD+ by Complex I). Electrons are then transferred from ubiquinol (oxidizing it back to ubiquinone) to cytochrome c via Complex III. Finally, electrons are transferred from cytochrome c to molecular oxygen via Complex IV, oxidizing cytochrome c and converting molecular oxygen to water.
The energy from these electron transfers is coupled (for Complexes I, III, and IV) to pumping protons against their electrochemical gradient from the mitochondrial matrix into the intermembrane space. The F1F0 ATP synthase (sometimes referred to as Complex V) harnesses the free energy released by protons flowing back down their electrochemical gradient to power the combination of ADP and inorganic phosphate to make ATP. This is the biochemical summary of oxidative phosphorylation, a process that is stoichiometrically equivalent to a combustion reaction (compounds containing carbon and hydrogen combining with molecular oxygen to liberate CO2, water, and energy).
Not only is this process the nexus of bioenergetics for most cell types, but oxidative phosphorylation is also intimately tied to regulation of apoptosis via cytochrome c. In addition, the mitochondrial electron transport system serves as one of the most important sources for damaging ROS. If the system works perfectly, molecular oxygen is completely reduced to water by Complex IV. However, at multiple points along the electron transport system, a single electron can be transferred to molecular oxygen, resulting in the formation of superoxide anion. Mitochondria are well equipped to deal with the constant hazard of superoxide. SOD2 (MnSOD) localizes to mitochondria and is present in concentrations that far exceed the normal concentrations of its substrate (one of the few known enzymes with this feature). SOD2 further catalyzes the already rapid dismutation of superoxide to molecular oxygen and hydrogen peroxide (Fig. 8.2). This serves to protect vulnerable lipids, proteins, and nucleic acids from oxidative damage. Indeed, SOD2 is such an important defense against baseline free radical injury that homozygous deletion of SOD2 is embryonically lethal in mice shortly after birth due to cardiomyopathy [63].
As with many other disease states, mitochondrial oxidative stress has been linked to the pathogenesis of PAH. Increased mitochondrial oxidant injury has been shown in genetic models of PAH, and driving mitochondrial free radical production enhances the development of PAH [64, 65]. Moreover, SOD2 has been shown to undergo epigenetic silencing in PAH, though the important consequence seems to be a loss of oxidative signaling from hydrogen peroxide as opposed to increased oxidant injury from superoxide. Restoring oxidative signaling by restoring SOD2 activity enhances appropriate apoptosis in the pulmonary vasculature and ameliorates the development of PAH [24].
The efficient transfer of electrons within the mitochondrial electron transport system and the proper function of antioxidant defenses require a close and tightly regulated physical relationship between the enzyme complexes and electron carriers. Disruption of the ultrastructure of mitochondria can lead directly to increased oxidant injury and an imbalance between apoptosis and proliferation. To help maintain mitochondrial ultrastructure in the face of normal and pathologic demands on bioenergetics and biosynthesis, and as a mechanism to help mitigate the effects of oxidant injury, mitochondria can combine together to form larger reticula (fusion) and can divide to form larger numbers of smaller mitochondria (fission) [66–70].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


