Chapter 24 Pulmonary Hypertension
Pulmonary hypertension and right ventricular dysfunction are common problems in the cardiothoracic intensive care unit (ICU). In this chapter the causes, implications, and treatment of pulmonary hypertension are discussed. The focus is on pulmonary hypertension that occurs in the context of underlying cardiac or pulmonary disease, rather than pulmonary hypertension that is idiopathic, familial, or associated with collagen vascular disease.
CLASSIFICATION AND PATHOPHYSIOLOGY
Definition and classification
The normal ranges for the systolic, diastolic, and mean pulmonary artery pressures are 20 to 30, 5 to 15, and 10 to 20 mmHg, respectively. Pulmonary vascular resistance index is normally 150 to 250 dyne.s.cm−5 (1.8 to 3.2 Wood units). The transpulmonary gradient is the difference between the mean pulmonary artery pressure and the left atrial pressure, and is normally 5 to 10 mmHg. The transpulmonary gradient is commonly used as a surrogate for pulmonary vascular resistance. If pulmonary vascular resistance is normal and left atrial pressure is greater than 5 mmHg, pulmonary artery diastolic pressure is about equal to left atrial pressure. The normal regulation of pulmonary vascular resistance is outlined in Chapter 1. Pulmonary arterial hypertension is defined as a sustained increase in mean pulmonary artery pressure above 25 mmHg at rest and 30 mmHg with exercise, with a pulmonary artery wedge pressure (PAWP) and left-ventricular end-diastolic pressure of less than 15 mmHg.1 However, in many patients with pulmonary hypertension secondary to cardiac disease, PAWP is (or has been) greater than 15 mmHg. The classification of pulmonary hypertension is shown in shown in Table 24-1.
Table 24-1 Revised World Health Organization Classification of Pulmonary Hypertension
Pulmonary hypertension associated with other conditions, including collagen vascular disease, congenital systemic-to-pulmonary shunts, HIV infection, drugs and toxins |
| Group 4: Pulmonary Hypertension due to Chronic Thrombotic or Embolic Disease |
| Group 5: Miscellaneous |
| Includes sarcoidosis, compression of pulmonary vessels, lymphangiomatosis, and histiocytosis X |
Modified from Simonneau G, Galie N, Rubin LJ, et al: Clinical classification of pulmonary hypertension. J Am Coll Cardiol 43:5S-12S, 2004. ASD, atrial septal defect; HIV, human immunodeficiency virus; PDA, patent ductus arteriosus; VSD, ventricular septal defect; COPD, chronic obstructive pulmonary disease.
Pathophysiology
In clinical practice, pulmonary arterial pressure is commonly assumed to be reflective of pulmonary vascular resistance, and in most circumstances this is the case. However, based on Equation 1-6, it is clear that pulmonary arterial pressure is also dependent on cardiac output and left atrial pressure. An increase in cardiac output can lead to an increase in pulmonary arterial pressure despite a fall in pulmonary vascular resistance. Conversely, an acute rise in pulmonary vascular resistance resulting in acute right ventricular failure and thus a fall in cardiac output may cause a fall in pulmonary arterial pressure.
The extent to which the right ventricle can cope with increased pulmonary vascular resistance depends on the time period over which the increased resistance has occurred. An untrained, thin-walled right ventricle is sensitive to acute rises in afterload. Thus, an abrupt rise in pulmonary vascular resistance (e.g., due to a massive pulmonary embolism) that requires a mean pulmonary arterial pressure of more than 40 mmHg to perfuse the pulmonary bed will rapidly lead to right ventricular failure. In contrast, if pulmonary vascular resistance increases gradually, right ventricular hypertrophy develops, which allows resting cardiac output to be maintained in the face of very high pulmonary vascular resistance (e.g., >20 Wood units). In this situation, pulmonary arterial pressure may approach or even exceed systemic arterial pressure.
With raised left atrial pressure, increased pulmonary arterial pressure can occur without raised pulmonary vascular resistance (see Equation 1-6). This is termed passive pulmonary hypertension. In passive pulmonary hypertension, the increase in pulmonary arterial pressure is usually mild and the transpulmonary gradient (see Chapter 1) is normal. The common causes of passive pulmonary hypertension are mitral valve disease and left ventricular dysfunction. The term active pulmonary hypertension denotes pulmonary hypertension due to vasoconstriction or anatomic restriction within the pulmonary bed in association with normal left atrial pressure. Pulmonary vascular resistance and transpulmonary gradient are elevated. Causes of active pulmonary hypertension include chronic lung disease and chronically elevated flow through the pulmonary circulation, such as that which occurs with uncorrected systemic-to-pulmonary shunts. The term reactive pulmonary hypertension refers to active pulmonary hypertension due to long-standing passive pulmonary hypertension. Chronically elevated left atrial pressure eventually causes pathologic changes within the pulmonary arterioles. The most common cause of reactive pulmonary hypertension in cardiac surgery patients is chronic mitral valve disease, particularly mitral stenosis. Rarely, left ventricular failure leads to reactive pulmonary hypertension. Pulmonary hypertension may be severe.
Pulmonary hypertension may be fixed or responsive. Changes in the pulmonary vascular bed that accompany active or reactive pulmonary hypertension include vasoconstriction, endothelial and smooth muscle proliferation, thrombosis, and fibrosis. Pulmonary hypertension that is associated with extensive thrombosis, fibrosis, or tissue destruction tends to be fixed, whereas pulmonary hypertension that is associated with abnormal vasoconstriction may be responsive to pulmonary vasodilating agents. Impaired production of vasodilator substances, such as nitric oxide, prostacyclin, and vasoactive intestinal peptide, and increased production of vasoconstrictor substances, such as endothelin-1 and thromboxane-A2, have been identified for a range of conditions associated with pulmonary hypertension.1 Alveolar hypoxemia is a potent stimulus for pulmonary vasoconstriction, and it contributes to pulmonary hypertension in patients with chronic lung disease.
DIAGNOSIS
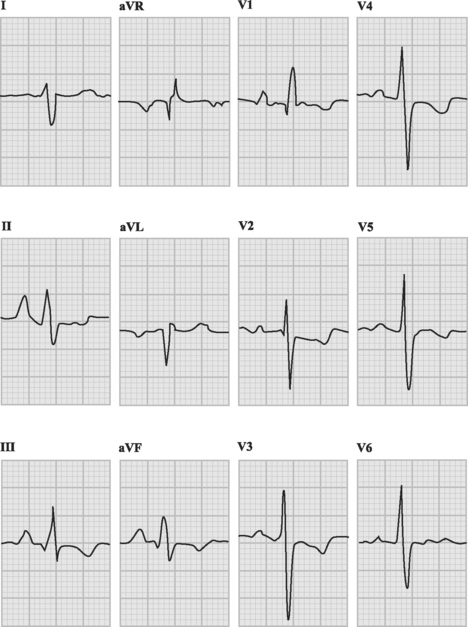
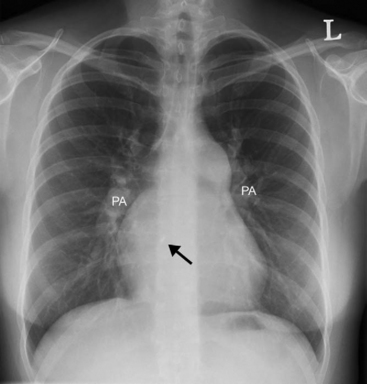
With severe pulmonary hypertension, the electrocardiogram (ECG) typically demonstrates right ventricular hypertrophy and right axis deviation (Fig. 24-1). On the chest radiograph, characteristic features of pulmonary hypertension include enlargement of the main and central hilar pulmonary arteries (Fig. 24-2) and reduced vascular markings in the peripheries (pruning of the vessels). Signs of left ventricular dysfunction or lung disease may be evident.
Pulmonary Artery Catheterization
Pulmonary artery catheterization may be used as part of a diagnostic workup (see Chapter 5) or in the ICU as a guide to management (see Chapter 8). Diagnostic right heart catheterization involves measurement of pulmonary arterial pressure, measurement of cardiac output, calculation of pulmonary vascular resistance, assessment of reversibility with pulmonary vasodilators (100% oxygen, nitroprusside, nitric oxide) and, in patients with intracardiac shunting, measurements of various oxygen saturations so as to allow estimation of the ratio of pulmonary to systemic flow (described in Chapter 5).
Echocardiography
With chronic pulmonary hypertension, the right ventricle is typically hypertrophied (wall thickness >0.5 cm and occasionally >1 cm) and dilated but, in the absence of ventricular failure, systolic function is preserved. Chronic right ventricular pressure overload leads to characteristic abnormalities in the shape and motion of the interventricular septum, which are described in Chapter 7
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


