Chapter 58
Pulmonary Hypertension
1. What is the hemodynamic criteria used to define pulmonary arterial hypertension?
2. What are the usual physical findings in patients with pulmonary hypertension?
The most common findings on physical examination include the following:
 Loud pulmonic valve closure sound (P2)
Loud pulmonic valve closure sound (P2)

 Murmur of tricuspid regurgitation (a systolic murmur over the left lower sternal border)
Murmur of tricuspid regurgitation (a systolic murmur over the left lower sternal border)
 Murmur of pulmonic insufficiency (a diastolic murmur over the left sternal border)
Murmur of pulmonic insufficiency (a diastolic murmur over the left sternal border)
 Jugular venous distension (indicating elevated central venous pressures)
Jugular venous distension (indicating elevated central venous pressures)





At the Fourth World Conference on Pulmonary Hypertension (held in Dana Point, Calif., in 2008), the term familial PAH was replaced by hereditary PAH, and schistosomiasis and chronic hemolytic anemia were added to Group 1 PAH. Current classification is outlined in Box 58-1. Pulmonary hypertension is classified as pulmonary arterial hypertension (Group 1), pulmonary hypertension resulting from left heart disease (Group 2), pulmonary hypertension resulting from lung diseases and/or hypoxia (Group 3), chronic thromboembolic pulmonary hypertension (CTEPH) (Group 4), and pulmonary hypertension with unclear multifactorial mechanisms (Group 5).
Box 58-1 Updated Pulmonary Hypertension Classification
Group 1. Pulmonary Arterial Hypertension (PAH)






















Group 4. Chronic Thromboembolic Pulmonary Hypertension (CTEPH)
Group 5. Pulmonary Hypertension with Unclear Multifactorial Mechanisms




From Dana Point, 2008. Simonneau G, Robbins I M, Beghetti M, et al: Updated clinical classification of pulmonary hypertension, J Am Coll Cardiol 54:S43–S54, 2009.
4. Is pulmonary hypertension a genetic disease?
About 6% of patients with PAH have hereditary PAH. Mutations in the gene encoding bone morphogenetic receptor 2 (BMPR2) were found in approximately 70% of families with hereditary PAH and in 20% to 30% of patients with idiopathic PAH. Due to incomplete penetrance, most patients with this mutation never develop the disease. A subject with a mutation has a 10% to 20% estimated lifetime risk of acquiring PAH.
5. What should the clinical evaluation for possible pulmonary hypertension include?
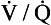 ) scan, hypercoagulable work-up (if indicated), electrocardiogram, and echocardiogram.
) scan, hypercoagulable work-up (if indicated), electrocardiogram, and echocardiogram.Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


