Pulmonary Hypertension
The diagnostic workup and treatment of pulmonary hypertension (PH) is part of cardiology as well as pulmonary practices and, due to different etiologies, is also sporadically encountered by rheumatologists, infectious disease specialists (HIV), and pediatricians. In general, PH (in particular, pulmonary arterial hypertension [PAH]) is a devastating disease, not only because it affects relatively young individuals but because the therapeutic options, although evolving, remain somewhat limited. In recent years, substantial progress has been made in unraveling the pathophysiology of PH directly resulting in some new medical therapies. Directions for diagnosis and treatment are formulated as an expert consensus document by ACCF/AHA1 and as guidelines by the ESC.2
DEFINITION AND CLASSIFICATION
PH is defined as mean pulmonary artery pressure (mPAP) >25 mm Hg at rest or >30 mm Hg with exercise. The latter component of this definition is questioned because it is not supported by published data, and healthy individuals can reach much higher values.3 PH encompasses a heterogeneous group of diseases with a common clinical manifestation. The true scope of the problem is not known, since many people have unrecognized PH. The terms “primary” (idiopathic or familial) and “secondary” PH have been abandoned and were replaced by a classification system adopted during the Second World Symposium on Pulmonary Hypertension in 1998 and most recently modified during the Fourth World Symposium in 2008 (Table 20.1).4 The aim of this clinical classification system is to group together different manifestations of disease sharing similarities in pathophysiologic mechanisms, clinical presentation, and therapeutic approaches. This makes sense because certain therapies for PAH are not effective in other forms of PH or might even be harmful. In this way, five major categories are outlined:
TABLE
20.1 Updated Clinical Classification of PH

Reprinted from Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. JACC 2009;54:43-54, with permission from Elsevier.
1. PAH
2. PH owing to left heart disease
3. PH owing to lung diseases and/or hypoxia
4. Chronic thromboembolic pulmonary hypertension (CTEPH)
5. PH with unclear multifactorial mechanisms
The terms PH, which is a hemodynamic and pathophysiologic condition, and PAH, a clinical condition, are thus not synonymous and one should pay attention to the specific population studied when interpreting trial results.
Pulmonary Arterial Hypertension
Although PAH probably only represents 4.2% of the total PH population, it has been the focus of attention since the first classification in 1973.5 Most recent data from registries estimate the prevalence around 15 to 50 cases per million adults.6,7 PAH is characterized by the presence of precapillary PH (pulmonary capillary wedge pressure [PCWP] < 15 mm Hg) in the absence of other causes of precapillary PH such as lung disease, pulmonary embolism, or other rare diseases (see Table 20.1). The nomenclature of the subgroups and the associated conditions has significantly evolved since 1973.
Idiopathic and familial PAHs (previously known as “primary pulmonary hypertension”) are rare diseases with a prevalence around six cases per million. Familial cases account for 5% to 10% of all PAH cases. Mutations in the bone morphogenetic protein receptor II (BMPR2) gene have been identified in at least 70% of patients with familial PAH and in 10% to 40% of patients with sporadic, idiopathic PAH.8,9 Lack of a functional BMPR2 gene appears to affect antiproliferative pathways of vascular cells. Genetic mutations in two other members of the transforming growth factor-β superfamily have also been identified, namely the activin receptor-like kinase 1 (ALK-1) and endoglin (ENG), which are associated with hereditary hemorrhagic teleangi- ectasia.8 Relatives of patients with familial idiopathic PAH should be advised about the availability of genetic testing and counseling in addition to echocardiographic screening.
PAH has been associated with the use of several drugs and toxins. Already in the 1960s, an association between anorexigens (appetite suppressant drugs that increase serotonin release and block serotonin reuptake) and PAH was observed following the introduction of aminorex fumarate.10 Subsequently in the 1980s, the same association was noticed after as little as 3 months of exposure to structurally related compounds such as (dex)fenfluramine.11
Several patient populations have a higher risk of developing PAH and are worth mentioning. Connective tissue diseases (CTDs), especially the limited cutaneous form of systemic sclerosis (formerly referred to as the CREST syndrome), are infrequently accompanied by PAH. The prevalence of hemodynamically proven PAH in systemic sclerosis is around 10% and can be the result of an isolated pulmonary arteriopathy as well as associated with interstitial fibrosis.12 In systemic lupus erythematosus, mixed CTD, rheumatoid arthritis, dermatomyositis, and Sjögren syndrome, PAH is observed to a lesser extent.
The incidence of PAH in patients infected with HIV is approximately 0.5%, which is still 6 to 12 times that of the general population. However, because of this low incidence, routine screening is not recommended.13 Cirrhosis patients with portal hypertension are another patient population with an increased incidence of PH (5% of patients referred for liver transplantation). This PH can be the result of a high flow state and a proliferative pulmonary arteriopathy with plexiform lesions, the so-called portopulmonary hypertension. The development of PH in this population is poorly understood and may portend poor survival (median 6 months) and high transplant mortality. Medical interventions and liver transplantation may sometimes reverse mild to moderate portopulmonary hypertension.14
Congenital heart disease is a well-recognized cause of PAH when the underlying systemic-to-pulmonary shunt is not corrected. It occurs most frequently with conditions where blood flow is high and the pulmonary vasculature is exposed to systemic level pressures (e.g., ventricular septal defect, patent ductus arteriosus). However, high flow only, as in atrial septal defect, can be sufficient. Once pulmonary vascular resistance (PVR) approaches or exceeds systemic vascular resistance, the shunt is reversed leading to desaturation and cyanosis (Eisenmenger syndrome). Although the pulmonary obstructive arteriopathy in this setting is identical to other PAH forms, prognosis is typically better.15
Pulmonary veno-occlusive disease (PVOD) and pulmonary capillary hemangiomatosis (PCH) are two rare disorders that share the histology of PAH but also demonstrate a number of differences and therefore remain somewhat difficult to classify. They are labeled as group 1′, a distinct category but not completely separated from PAH. They exhibit the findings of pulmonary venous hypertension including pulmonary hemosiderosis, interstitial edema, and lymphatic dilatation.
Pulmonary Hypertension Owing to Left Heart Disease
In this category, pathology is situated on the left side of the heart and PH is the result of backward transmission of the pressure elevation (postcapillary passive). In these circumstances, transpulmonary pressure gradient (TPG) and PVR are within normal limits. However, in other circumstances, PAP elevation is greater than PCWP, reflecting an increased TPG and PVR (postcapillary reactive or “out of proportion” PH). The latter can be due to increased vasomotor tone or to fixed structural obstructive remodeling of the pulmonary artery vessels. Some of the newer medical therapies first tested in PAH are now also tested in this “out of proportion” patient population.
Pulmonary Hypertension Owing to Lung Diseases and/or Hypoxia
Group 3 PH encompasses respiratory disease such as chronic obstructive pulmonary disease (COPD), interstitial lung disease, and sleep disordered breathing. These diseases can cause hypoxic vasoconctriction, mechanical stress of hyper- inflated lungs, loss of capillaries, and inflammation.
Chronic Thromboembolic Pulmonary Hypertension
In line with the reasoning behind the present classification, CTEPH forms a different group. Most of the time, it is caused by emboli obstructing pulmonary arteries, but, interestingly, in the nonoccluded areas, a pulmonary arteriopathy indistinguishable from that of PAH can develop.16 There is accumulating evidence that CTEPH may also develop in the absence of previous pulmonary embolism, being the result of local thrombotic or inflammatory lesions in the pulmonary vasculature.17
Pulmonary Hypertension with Unclear Multifactorial Mechanisms
This group comprises a heterogeneous collection of diseases with uncertain pathogenetic mechanisms and pathologic pictures leading to PH.
PATHOPHYSIOLOGY
Applying Ohm’s law to the pulmonary circulation results in pressure difference (mPAP − PCWP) = flow (cardiac output [CO]) x resistance (PVR). Formulated like this, elevation of the mPAP must be the consequence of elevation in PCWP, increase in flow, or increase in PVR. However, the capacity of the pulmonary circulation can be increased by both recruitment and distention resulting in lower PVR. This means that flow can increase substantially without an appreciable change in PAPs in physiologic conditions. These low pressure, low resistance, and high compliance characteristics of the pulmonary vascular bed are regulated by a balance of vasodilators/vasoconstrictors and cell proliferation/apoptosis. Various external and host genetic factors may disturb this intricate balance resulting in excessive vasoconstriction, vascular remodeling, and thrombosis leading to pulmonary (arterial) hypertension (Fig. 20.1). Although it is often not clear what exact processes initiate the pathologic changes seen, substantial progress has been made in our understanding of the various biochemical pathways and cell types involved. Like cancer and atherosclerosis, PAH does not have a single cause: a “multihit model” is more likely.18
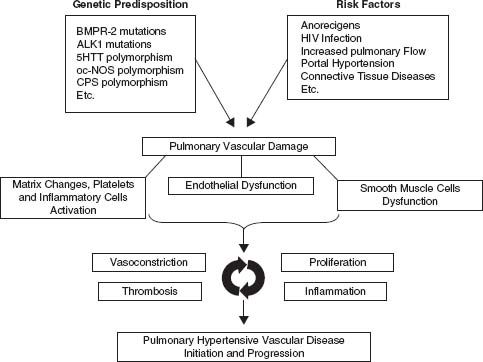
FIGURE 20.1 Pathophysiology of PH. (Galié N, Torbicki A, Barst R, et al. Guidelines on diagnosis and treatment of pulmonary arterial hypertension: the Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J. 2004;25:2243–2278, by permission of Oxford University Press.) BMPR-2, bone morphogenetic protein receptor II; ALK-1, activin receptor-like kinase 1; 5HTT, serotonin transporter; ec-NOS, endothelial cell nitric oxide synthase; CPS,carbamyl-phosphate synthase; HIV, human immunodeficiency virus.
Prostacyclin and Thromboxane A2
Both prostacyclin and thromboxane A2 are major arachidonic acid metabolites of vascular cells. Prostacyclin is known for its potent vasodilating, antiproliferative, and platelet-inhibiting properties, whereas thromboxane A2 does the opposite. Typically, in PAH, the balance is shifted toward thromboxane A2 as assessed by decreased urinary levels of a prostacyclin metabolite, increased urinary thromboxane B2 levels, and decreased expression of prostacyclin synthase in small- and medium-sized pulmonary arteries.19,20
Endothelin-1
Endothelin-1 (ET-1) belongs to a family of vasoconstrictor peptides that plays an important role in vascular control. It is produced by endothelial cells and secreted mainly from the abluminal side toward the adjacent vascular smooth muscle cells. Two types of receptors exist: endothelin receptor A (ETA), expressed on vascular smooth muscle cells, and endothelin receptor B (ETB), expressed on both vascular endothelial cells and smooth muscle cells. Stimulation of both receptors on the vascular smooth muscle cells causes vasoconstriction and has a mitogenic effect, whereas stimulation of the ETB on the endothelial cells causes vasodilatation via increased production of prostacyclin and nitric oxide (NO). In patients with PH, ET-1 levels are often increased, and ET-A receptors are abundant.21
Nitric Oxide
NO is produced in endothelial and epithelial cells in the lung from L-arginine by three isoforms of nitric oxide synthases (NOS). Once formed, the effects of NO are mediated by cyclic guanosine monophosphate (cGMP), which is rapidly inactivated by the phosphodiesterase enzymes, especially type 5 (PDE-5). Decreased endothelial NOS (NOS 3) has been observed in PAH patients. NO is a potent vasodilator, an inhibitor of platelet activation and of vascular smooth muscle cell proliferation. These properties render it an excellent target for therapy (Fig. 20.2).22
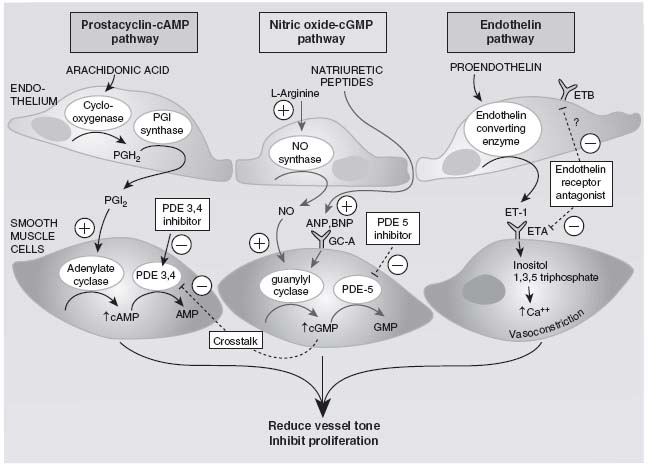
FIGURE 20.2 Targets for current or emerging therapies in PH. (Adapted from Humbert M, Sitbon O, Simonneau G, et al. Treatment of pulmonary hypertension. N Enal J Med. 2004;351:1425–1436.)
Other Vascular Effectors
Serotonin (5-hydroxytryptamine) is also a vasoconstrictor that promotes smooth muscle cell hypertrophy and hyperplasia. Serotonin transporter overexpression has been associated with PH.23 Vasoactive intestinal peptide (VIP) has a pharmacologic profile similar to prostacyclins and levels are decreased in PAH patients.24 Angiopoietin-1, an angiogenic factor essential for vascular lung development, seems to be upregulated in cases of PH, correlating directly with the severity of the disease. In addition, inflammatory cells are ubiquitous in PAH and can further cause pulmonary vascular damage and endothelial dysfunction by cytokine release and cytotoxic effects.
Hemodynamic Consequences
The previously mentioned vasoconstriction, vascular smooth muscle cell proliferation, and thrombosis lead to an elevated PVR and an increase in right ventricular (RV) afterload. This, on its terms, results in RV dilatation and increased free wall tension, leading to hypertrophy. The ability of the right ventricle to compensate and preserve CO is crucial for the further evolution of this disease. Once symptoms of RV failure emerge, manifested as further dilatation, thinning of the wall, and tricuspid regurgitation, prognosis is poor.
DIAGNOSIS AND EVALUATION
Clinical Evaluation
The symptoms of PH are usually gradual in onset and nonspecific explaining why the lag time between symptom onset and diagnosis approaches 2 years in 90% of PAH patients. These symptoms include dyspnea on exertion, fatigue, weakness, chest pain, palpitations, syncope, abdominal distention, and pedal edema. Clinical examination of PH may reveal a left parasternal lift, a loud P2 at apex, a pan-systolic murmur of tricuspid regurgitation that increases with inspiration, a diastolic murmur of pulmonary insufficiency, and a RV S3. Jugular vein distension, hepatomegaly with a pulsatile liver, peripheral edema, and ascites are often indicative of advanced stages with frank right-sided heart failure.
At least in certain populations at risk, the careful clinician will suspect the diagnosis whenever the previously mentioned symptoms or signs occur. These populations include:
1. Known BMPR2 mutation
2. First-degree relative of patient with BMPR2 mutation or within pedigree of two or more patients with PAH
3. Systemic sclerosis
4. Sickle cell disease
5. HIV infection
6. Portal hypertension
7. Prior appetite suppressant use
8. Congenital heart disease with shunt
9. Recent acute pulmonary embolism
10. Left heart disease
11. COPD, interstitial lung disease, or sleep apnea
In the first four categories, yearly echocardiographic screening is recommended.1
Echocardiography
If PH is suspected based on the history, risk factor assessment, and physical examination, an echocardiogram is the next appropriate study. By using the Doppler technique, peak velocity of the tricuspid regurgitation jet can be measured. From this measured velocity, the pressure difference between right ventricle and right atrium can be estimated on the basis of the simplified Bernoulli equation ( P = 4ν2). On the condition that there is no pulmonic valve stenosis, pulmonary artery systolic pressure (PASP) = 4 x (tricuspid regurgitation velocity)2 + right atrial pressure (RAP). RAP can be estimated on the base of inferior vena cava characteristics.
P = 4ν2). On the condition that there is no pulmonic valve stenosis, pulmonary artery systolic pressure (PASP) = 4 x (tricuspid regurgitation velocity)2 + right atrial pressure (RAP). RAP can be estimated on the base of inferior vena cava characteristics.
Other two-dimensional echocardiographic characteristics might raise or reinforce suspicion of PH; for example, right atrial or RV dilatation, flattened interventricular septum with D-shaped left ventricle, increased RV wall thickness, dilatation of the pulmonary artery, and pericardial effusion. These features tend to occur later in the course of the disease. Echocardiography is also helpful in detecting and characterizing left heart disease and congenital cardiac abnormalities.
Although echocardiography is a useful screening tool, Doppler-derived pressure estimation can both underestimate PASP in patients with severe tricuspid regurgitation and overestimate PASP in non-PH patients. Ultimate confirmation should come from right heart catheterization (RHC).
Other (Imaging) Studies
In typical cases of PH, the ECG reflects RA dilatation, RV hypertrophy with strain, and QRS complex frontal plane right axis deviation. In advanced stages of the disease, atrial flutter or atrial fibrillation often occur leading to further clinical deterioration.
Chest x-rays often reveal central pulmonary arterial dilatation with “pruning” (loss) of the peripheral blood vessels, clear lung fields, and a prominent RV border. Chest computed tomography (CT) and ventilation/perfusion (V/Q) scans are indicated to exclude primary parenchymal or thromboembolic diseases as a cause of PH. For excluding thromboembolic disease, V/Q scan is the preferred screening test. A normal or very low probability scan virtually excludes CTEPH, while a high probability scan warrants further evaluation with a pulmonary angiogram. Pulmonary angioscopy is sometimes performed in specialized centers in cases of chronic thromboembolic PH to determine surgical candidacy.
Hemodynamic Evaluation
RHC is required to confirm the diagnosis of PH, to assess the etiology and severity, to test for vasoreactivity of the pulmonary circulation, and in the follow-up of treatment.25 When performed at experienced centers, morbidity (1.1%) and mortality (0.055%) rates are low.26 Consecutively, RAP, right ventricular pressure (RVP), PAP, and PCWP are recorded using a balloon-tipped fluid-filled catheter. CO can be determined by using the thermodilution method and/or the Fick method (measurement of mixed venous saturation SvO2 needed). The PCWP is supposed to reflect left atrial pressure (LAP) and ultimately, in the absence of mitral stenosis, left ventricular end diastolic pressure (LVEDP). This measurement is very important because it helps differentiating PH associated with left heart disease from other conditions. It is, however, the pressure that is most subject to error in measurement and interpretation. There are several ways to confirm catheter position:
1. Stable fluoroscopic position
2. Presence of highly oxygenated (>95%) blood when a sample is drown from the distal port
3. Stagnation of injected dye in the pulmonary artery



