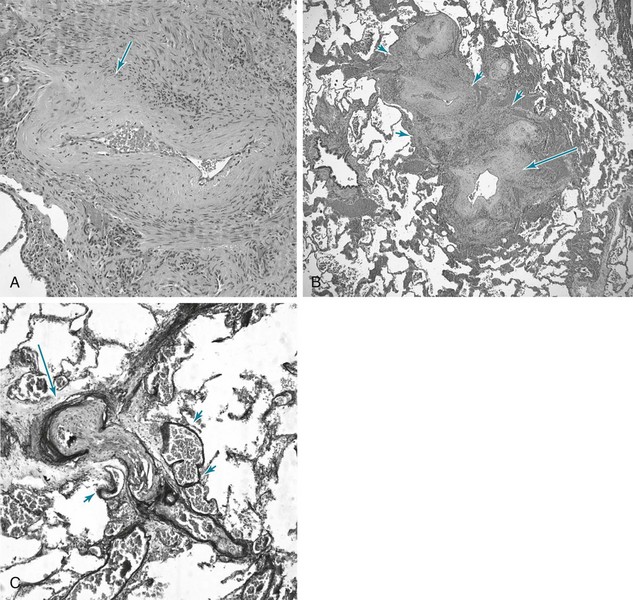
14 SPECIFIC DISORDERS ASSOCIATED WITH PULMONARY HYPERTENSION Idiopathic Pulmonary Arterial Hypertension and Related Disorders (Group 1 PAH) Pulmonary Hypertension Owing to Left Heart Disease (Group 2 PH) Pulmonary Hypertension Owing to Lung Disease and/or Hypoxia (Group 3 PH) Chronic Thromboembolic Pulmonary Hypertension (Group 4 PH) Pulmonary Hypertension with Unclear Multifactorial Mechanisms (Group 5 PH) The current classification for clinical categories of pulmonary hypertension is summarized in Table 14-1. Clarification of a few points is pertinent. First, the term pulmonary hypertension (PH) simply refers to elevated pulmonary arterial pressure, which may be due to a number of different mechanisms. The term pulmonary arterial hypertension (PAH) is reserved for specific types of PH—those categorized under Group 1 in the classification system in Table 14-1. Elevation of pulmonary arterial pressure may be acute or chronic and either reversible or irreversible, depending on the causative factors. In some cases, chronic PH is punctuated by further acute elevations in pressure, often as a result of exacerbations of the underlying disease. Second, the development of right ventricular hypertrophy is the consequence of chronic PH, whatever the primary cause of the latter. When PH is due to disorders of any part of the respiratory apparatus (airways, parenchyma and blood vessels, chest wall, respiratory musculature, or central nervous system controller), the term cor pulmonale is used to refer to the resulting right ventricular hypertrophy. This term should not be used to describe the right ventricular changes occurring as a consequence of primary cardiac disease or increased flow to the pulmonary vascular bed. Table 14-1 UPDATED CLINICAL CLASSIFICATION OF PULMONARY HYPERTENSION (DANA POINT, 2008) 1. PULMONARY ARTERIAL HYPERTENSION (PAH) 1.5. Persistent pulmonary hypertension of the newborn 1′ Pulmonary Veno-Occlusive Disease (Pvod) and/or Pulmonary Capillary Hemangiomatosis (Pch) 2. PULMONARY HYPERTENSION OWING TO LEFT HEART DISEASE 3. PULMONARY HYPERTENSION OWING TO LUNG DISEASE AND/OR HYPOXIA 3.1. Chronic obstructive pulmonary disease 3.2. Interstitial lung disease 3.3. Other pulmonary diseases with mixed restrictive and obstructive pattern 3.4. Sleep-disordered breathing 3.5. Alveolar hypoventilation disorders 3.6. Chronic exposure to high altitude 4. CHRONIC THROMBOEMBOLIC PULMONARY HYPERTENSION (CTEPH) 5. PULMONARY HYPERTENSION WITH UNCLEAR MULTIFACTORIAL MECHANISMS 5.1. Hematologic disorders: myeloproliferative disorders, splenectomy 5.2. Systemic disorders: sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, neurofibromatosis, vasculitis 5.3. Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders 5.4. Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure on dialysis Modified from Simonneau G, Robbins IM, Beghetti M, et al: Updated clinical classification of pulmonary hypertension, J Am Coll Cardiol 54(Suppl 1):S43–S54, 2009. A number of factors contribute to the pathogenesis of PH, both acutely and chronically. First, occlusion of a sufficient cross-sectional area of the pulmonary arteries by material (e.g., pulmonary emboli) within the vessels is an important factor (discussed in Chapter 13). In acute embolism, in which massive pulmonary emboli occlude more than half to two thirds of the vasculature, pulmonary arterial pressure is elevated. The right ventricle may dilate in response to its acutely increased workload because of insufficient time for hypertrophy to occur. In contrast, in chronic embolic disease, multiple and recurrent pulmonary emboli may elevate pulmonary arterial pressures during a period sufficient for right ventricular hypertrophy to occur. Second, remodeling of the pulmonary arterial walls causing diminution of cross-sectional area is a potential factor. Disorders acting by this mechanism are characterized by intimal and medial changes (see Pathology) that lead to thickening of the arterial and arteriolar walls and narrowing or obliteration of the lumen. This group of disorders with pulmonary arterial pathology includes idiopathic pulmonary arterial hypertension (IPAH, formerly called primary pulmonary hypertension). The familial form of this condition, called heritable pulmonary arterial hypertension, in most cases is related to mutations of the gene on chromosome 2 that encodes the bone morphogenetic protein receptor type 2 (BMPR2). Abnormalities in this receptor are believed to lead to dysregulation of proliferative responses in the endothelium and pulmonary arterial smooth muscle cells, producing the well-described pathologic changes in small pulmonary arteries and arterioles (again, see Pathology). Lesions pathologically similar to those seen in IPAH are also observed in other conditions associated with PAH (e.g., scleroderma, portal hypertension, human immunodeficiency virus [HIV] infection) or with exposure to drugs and toxins (e.g., cocaine, methamphetamine, certain diet drugs). When compromise of the pulmonary vasculature and increased resistance to flow are sufficiently pronounced in these primary disorders of the vessel wall, the level of PH can be quite severe, both at rest and with exercise. A fourth mechanism of PH is vasoconstriction in response to hypoxia and, to a lesser extent, to acidosis. The importance of this mechanism is related to its potential reversibility when normal PO2 and pH values are restored. In several causes of cor pulmonale, particularly chronic obstructive pulmonary disease (COPD), hypoxia is the single most important factor leading to PH and is potentially the most treatable. Acidosis, either respiratory or metabolic, causes pulmonary vasoconstriction and, although it is less important than hypoxia, may augment the vasoconstrictive response to hypoxia (discussed in Chapter 12). Although PH is classified into different clinical categories (see Table 14-1), as the disease progresses and remodeling occurs, the pathologic findings in the pulmonary arteries of patients with PH are similar regardless of the underlying cause. This section focuses on these general changes, which are particularly well illustrated in the lungs of patients with IPAH. The most prominent abnormalities are seen in pulmonary arterial tree vessels with a diameter of less than 1 mm: the small muscular arteries (0.1–1 mm) and the arterioles (<0.1 mm). The muscular arteries show hypertrophy of the media, composed of smooth muscle, and hyperplasia of the endothelial cells that make up the intimal layer lining the vessel lumen. In the arterioles, a significant muscular component to the vessel wall is not normally present, but with PH, these vessels undergo “neomuscularization” of their walls (Fig. 14-1, A). In addition, the arteriolar intima proliferates. As a result of medial hypertrophy and encroachment of proliferating endothelial cells into the vessel, the luminal diameter is significantly decreased, and the pulmonary vascular resistance is elevated. Ultimately, the lumen may be completely obliterated and the overall number of small vessels greatly diminished. In some cases of severe PH, particularly when due to IPAH or secondary to congenital intracardiac shunts, cells originating in the vessel wall (smooth muscle cells, endothelial cells, and fibroblasts) will form so-called plexiform lesions, appearing as a plexus of small, slitlike vascular channels (Fig. 14-1, B). Although the pathogenesis of these lesions is not precisely understood, disordered endothelial cell growth has been documented in patients with IPAH. It appears likely that the endothelial cells in many patients with severe PH have acquired a dysfunctional pro-proliferative phenotype that is resistant to apoptosis (cell death). When PH becomes marked, other changes are commonly seen in the larger (elastic) pulmonary arteries (Fig. 14-1, C
Pulmonary Hypertension
Pathogenesis
Pathology
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Thoracic Key
Fastest Thoracic Insight Engine