Chapter 58 Pulmonary Hypertension
Classification
Pulmonary hypertension was previously classified as either primary or secondary, depending on the absence or the presence, respectively, of identifiable causes or risk factors. The diagnosis of primary pulmonary hypertension was one of exclusion after ruling out all other causes for PH. Subsequent classification schemes have attempted to create categories of PH that share pathologic and clinical features, as well as similar therapeutic options. These classification schemes have allowed investigators to conduct clinical trials in well-defined patient groups with a shared underlying pathogenesis for their PH, resulting in the development of new targeted drug therapies; consequently, improvements in both quality of life and survival can now be expected in this otherwise deadly disease. This more inclusive category of PAH also has afforded increased opportunities for treatment of some less common forms of the disorder that were previously too rare for individual treatment studies. The most recent classification scheme was the product of the aforementioned Fourth World Symposium on Pulmonary Hypertension (Box 58-1).
Box 58-1
Updated Clinical Classification of Pulmonary Hypertension
Group 1: Pulmonary Arterial Hypertension
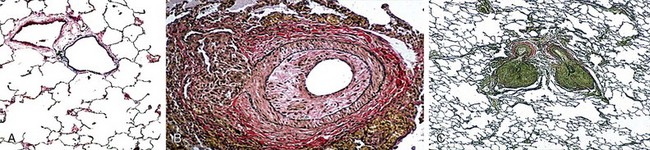
PAH is a subset of PH defined as a mPAP greater than 25 mm Hg determined with the patient at rest and a normal pulmonary capillary wedge pressure (PCWP) and/or left ventricular end-diastolic pressure (LVEDP) and a lesion localized to the pulmonary arteriole (Figure 58-1, A to C). Unfortunately, a limitation of these classification schemes is the fact that many of these patients have “multifactorial pulmonary hypertension.” The clinician is thus faced with treating PH in a variety of clinical scenarios that often include features from more than one of the World Health Organization (WHO) classification categories (i.e., groups 1 to 5, with an additional 1′ grouping as described later on). For example, the clinical presentation may include somewhat elevated pulmonary venous pressures, mild to moderate obstructive or restrictive lung disease, or a form of valvular heart disease that typically would not account for pulmonary hypertension severity. Patients with such “out of proportion” PH are not included in clinical trials; therefore, data pertaining to the safety and efficacy of conventional PAH therapies in this population are extremely limited.
Heritable Pulmonary Arterial Hypertension
Sporadic mutations in BMPR2 have been identified in approximately 11% to 40% of patients with presumably the idiopathic form of PAH and are seen in 70% to 80% of patients with familial PAH but are relatively uncommon in patients with so-called associated PAH (i.e., category 1.4 in Box 58-1). Although penetrance is low and only approximately 25% of carriers will go on to develop PAH, genetic anticipation also has been demonstrated (i.e., in affected families, each successive generation has more severe PAH developing at an earlier age). BMPR2 has been localized to chromosomal region 2q31-32, and inheritance occurs in an autosomal dominant fashion. Recently, it has been suggested that patients with PAH associated with BMPR2 mutations may represent a subgroup with more severe disease who are less likely to demonstrate vasoreactivity than those with IPAH. Because this mutation can occur sporadically in as many as 25% of patients with PAH and does not occur in all patients with so-called familial PAH, the term heritable is now favored over the designation familial.
Drug- and Toxin-Induced Pulmonary Arterial Hypertension
A number of risk factors for the development of PAH have been identified (Box 58-2). Risk factors for PAH include “any factor or condition that is suspected to play a predisposing or facilitating role in the development of the disease.” Such risk factors have been categorized as “definite, very likely, possible, or unlikely, based on the strength of their association with [pulmonary hypertension] and their probable causal role.” As a result of the Dana Point symposium, methamphetamine use was reclassified as a very likely risk factor for the development of PAH.



