(1)
Department of Cardiothoracic Surgery, Papworth Hospital, Cambridge, UK
(2)
The University of New South Wales, medical School, Sydney, NSW, Australia
9.1 Introduction
Failure of the right ventricle (RV) can occur in both the acute and chronic thromboembolic disease states. A pulmonary embolus refers to an obstruction in the pulmonary arterial vasculature. Most commonly, this material comprises of thrombus as a form of venous thromboembolism (VTE) but also includes tumour, fat and air. Pulmonary emboli usually present acutely with symptoms secondary to a sudden rise in pulmonary vascular resistance (PVR) and RV dilatation when large. Pulmonary embolism can also lead to chronic thromboembolic pulmonary hypertension (CTEPH), and in this situation the RV has time to adapt to the increasing PVR and hypertrophy occurs as well as dilatation. CTEPH can be treated with a surgical procedure to remove the pulmonary artery obstruction – pulmonary endarterectomy (PEA), with a dramatic reduction in PVR postoperatively. This gives a unique opportunity to observe remodelling of the RV as it recovers following surgery.
9.2 Acute Pulmonary Emboli
It is difficult to be certain of the true incidence as some emboli may be asymptomatic. In Europe it has been estimated to effect 40 per 100,000 population. Pulmonary emboli (PE) have been shown to be a major cause of mortality in the developed world. In Europe alone it has been reported to account for 300,000 deaths annually [1]. However, with advances in early recognition, diagnosis and treatment, particularly in the fields of imaging and anticoagulation, the mortality from PE has steadily declined [2]. Prophylaxis in patients undergoing surgery has also contributed to a reduction in incidence. There is a higher incidence among certain populations, particularly those that are predisposed to prolonged periods of immobility – hospitalised patients, stroke patients or patients having undergone recent surgery. Patients who have known thrombotic disorders and known malignancies and women during pregnancy have also been seen to have a higher incidence of PEs [3, 4]. The association with air travel is probably overstated.
Virchow’s triad underpins the contributing factors towards venous thromboembolism – alterations in blood flow (i.e. stasis), vascular endothelial injury and alterations in the constituents of the blood (i.e. inherited or acquired hypercoagulable state). Deep venous thrombosis (DVT) usually occurs in the lower limb and embolism results in thrombus getting trapped in the pulmonary arteries [6].
9.2.1 Right Ventricular Failure and Acute Pulmonary Embolism
The impact of PE is predominantly determined by the size. PE has been classified into massive or non-massive determined by the presence of shock (systemic systolic blood pressure of <90 mmHg). Within the non-massive group, there is a subset of patients with RV dysfunction who are not in shock; these patients are sometimes classified as sub-massive. In the setting of a large acute PE, the RV is subject to a significant change in haemodynamics. As described in the Pathophysiology chapter, the RV is not equipped to cope with these sudden changes and hence it is no surprise that failure of RV follows such an event.
In individuals without pre-existing pulmonary arterial hypertension (PAH), it has been reported that 25–30 % of the pulmonary vasculature needs to be obstructed to cause any rise in pulmonary artery pressures [7] with an obstruction of greater than 50 % to precipitate acute right heart failure. This group of patients do not have the compensatory mechanisms that may have developed as a result of chronic disease (i.e. RV hypertrophy) and are more susceptible to RV failure from acute PE. However, the size of the clot is not the only factor in determining the extent of the physiologic response to acute PE as this is also determined by the release of vasoactive mediators as a result of the acute hypoxic injury. Mediators such as serotonin, thromboxane and histamine are released as a result of the platelet- and thrombin-rich clots. The resulting vasoconstriction further increases pulmonary vascular resistance; this adds to the increase in afterload secondary to the pulmonary artery obstruction itself.
The fact that RV failure is the cause for circulatory collapse following pulmonary artery obstruction was first put forward in 1936 [8]. The presence of RV dysfunction is an important prognostic indicator following PE. Massive PE (with shock) is associated with a high mortality and this is even higher when cardiopulmonary resuscitation is required [9]. However, only a minority of patients with PE develop cardiogenic shock in the acute setting. A degree of RV dysfunction however is more common, even in normotensive patients after PE, and has been observed on echocardiography in up to 70 % of cases [10]. Evidence of RV dysfunction is important; it can more than double the short-term mortality when compared to patients without any evidence of RV dysfunction. It is important to have a high index of suspicion for RV dysfunction, as often it is a progressive feature with significant deterioration up to 48 h after the initial presentation. Furthermore, patients with PE secondary to DVT may have a continued embolic load that may result in the acute decline of the clinical state [11].
9.2.2 Clinical Presentation
In the most severe cases of massive PE, patients present with syncope, cardiogenic shock or even cardiac arrest. In patients where the presentation is not as dramatic, it is even more pertinent to maintain a high index of suspicion and look for subtle signs of impeding RV failure. Clinical findings are typical for someone with right-sided volume overload or chronic PAH with elevated jugular venous pressure, in particular, the v wave may be more pronounced. In an attempt to compensate for the increase in afterload, the increased pressure on the RV may result in a parasternal heave on palpation. This is often associated with a loud second heart sound and in some cases a fourth heart sound may also be auscultated. Once tricuspid regurgitation develops a pansystolic murmur is audible at the left sternal edge and a large C-V wave is visible in the jugular venous pulse. In patients with a patent foramen ovale (PFO), the rise in right atrial pressure can reverse flow, with right to left shunting. This can worsen hypoxaemia in acute PE. If any further embolic thrombus arrives in the right atrium a paradoxical embolism can occur with passage of thrombus through the PFO to the left atrium with the risk of stroke (Fig. 9.1).
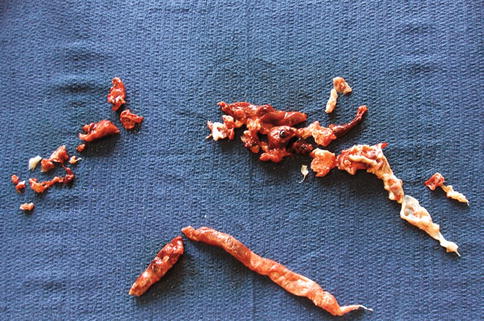
Fig. 9.1
Subacute pulmonary embolus removed at embolectomy with impending paradoxical embolism traversing a patent foramen ovale
9.2.3 Investigation
Electrocardiograms usually show patterns of right heart strain, repolarisation abnormalities or even signs of ischaemia. These include sinus tachycardia, complete or incomplete right bundle branch block, right axis deviation or an S1Q3T3 pattern (which is characteristic of PE). QR in V1 and T wave inversion in V2 have been shown to be related to RV dysfunction with specificity of 97 % and 94 %, respectively [12]. It has also been demonstrated that patients with atrial fibrillation, low-voltage QRS complexes and premature ventricular complexes as a result of an acute PE have a higher 30-day mortality [13]. Patients with PE frequently have elevated levels of troponin and brain natriuretic peptide (BNP). These markers have been shown to be linked with the degree of RV dysfunction and in turn predictors of associated mortality. Troponin has been shown to be raised in patients with RV dysfunction; this is due to myocardial ischaemia resulting from the effects of the pulmonary embolism [14]. Myocardial stretch as result of dilatation of the RV with increasing afterload is associated with release of BNP. BNP in turn correlates to increased mortality in the setting of acute PE [15, 16].
As previously mentioned, RV dysfunction in pulmonary emboli is associated with higher mortality and therefore echocardiography should always be considered, even in normotensive patients. Echocardiographic findings are likely to be seen in cases where there is greater than 30 % pulmonary artery obstruction [17]. In contrast to the global hypokinesis of the RV seen in patients with PAH, acute PE-driven RV dysfunction is characterised by sparing the apex of the RV, and the most significant hypokinesis is seen in the free wall and base. This clinical finding is referred to as ‘McConnell sign’ and has been shown to have a high specificity for diagnosing pulmonary emboli [17].
Dilatation of the RV is assessed by measuring the absolute end-diastolic diameter, as well as the ratio between the end-diastolic diameters of the right and left ventricle (LV). A value of >30 mm or a ratio >1 will signify RV dilation. Measurements of the RV wall thickness are useful as values greater than 5 mm are suggestive of some coexisting, likely chronic, cardiopulmonary disease. As analysed in echocardiography chapter, changes in geometry of the ventricles in RV dilation, whereby there is bowing of the septum towards the LV, result in altered diastolic flow across the mitral valve. The Doppler wave across the mitral inflow consists of a passive atrial filling wave (E wave) and an atrial systolic filling wave (A wave). When the concentric LV becomes ‘D shaped’ as a result of the bowing septum, there is an increased A wave; this is referred to as ‘E to A reversal’ (see Fig. 9.2). As a bedside investigation, echocardiography is also useful in establishing the diagnosis of PE, as emboli in the RV, inferior vena cava or main pulmonary arteries may be identified.
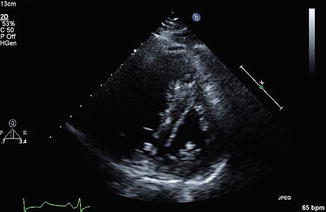
Fig. 9.2
Echocardiogram demonstrating the ‘D’-shaped left ventricle with flattened intraventricular septum
9.2.4 Treatment
The main treatment remains anticoagulation, initially with heparin, followed by a vitamin K vantagonist. For patients with massive PE, immediate thrombolysis is recommended in all guidelines and reduces the risk of death, but with a risk of haemorrhagic complications of up to 20 % in some series. Emergency surgical embolectomy or catheter-based therapy are alternatives as second-line therapy. Supportive treatments include inotropic agents and optimisation of preload. In the most severe cases, extracorporeal membrane oxygenation (ECMO) may be indicated.
9.3 Chronic Thromboembolic Pulmonary Hypertension
9.3.1 Epidemiology and Aetiology
Chronic thromboembolic pulmonary hypertension (CTEPH) is a progressive, life-threatening form of precapillary PAH that may be amenable to cure by pulmonary endarterectomy (PEA) surgery. CTEPH develops as a result of obstruction and/or occlusion of branches of the pulmonary arterial tree following incomplete resolution of a single or recurrent episode of VTE [18]. The subsequent fibrosis of obstructive thrombus material and secondary pulmonary arterial remodelling lead to an increased PVR, PAH and progressive right heart failure which cause significant morbidity and mortality in affected patients.
The exact prevalence of CTEPH is unknown with a recent prospective study of UK pulmonary hypertension centres reporting an incidence of approximately 1.8 cases per million [19]. Symptomatic CTEPH has been found to affect approximately 4 % of patients within 2 years of first symptomatic PE [20]. The disease is, however, considered to be widely under-diagnosed due to non-specific presentation symptoms, its variable association with PE and difficulty in identification of CTEPH on diagnostic imaging at low volume centres. Significantly, 25–63 % of CTEPH cases present with no preceding history acute PE [21, 22]. Given the incidence of venous thromboembolism including PE [23], the true incidence of CTEPH is, therefore, likely to be much greater than reported.
Impaction and occlusion of the pulmonary arterial vascular tree by acute VTE is the precipitating factor in CTEPH. Even following timely diagnosis and management of PE with anticoagulation, patients with CTEPH fail to achieve complete thrombus resolution through normal physiological mechanisms. Moser and Braunwald in 1973 first proposed the concept of CTEPH as a ‘dual compartment’ vascular disorder consisting of both proximal pulmonary vascular obstruction and a distal small vessel arteriopathy [24].
9.3.2 Right Ventricular Failure in CTEPH
Right heart failure in CTEPH is primarily a result of exposure of the RV to chronic elevation of afterload with pressure and volume overload, as analysed in the Pathophysiology chapter. Ongoing pulmonary vascular obstruction results in adaptive wall hypertrophy of the RV. Sustained pressure overload in CTEPH, however, leads to progressive loss of RV contractility and eventual dilatation which allows for compensatory preload and maintenance of RV stroke volume despite reduced fractional shortening. The increased wall stress that occurs in dilatation leads to progressively increased myocardial oxygen demand and decreased RV perfusion, giving rise to a vicious cycle of ongoing impaired contractility and dilatation (see Fig. 9.3). The specific mechanism by which the RV transitions from hypertrophy to dilatation secondary to PH is unknown [25, 26].
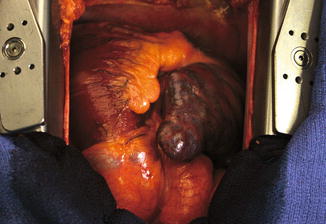
Fig. 9.3
Surgical view of the failing right heart in severe pulmonary arterial hypertension
Pulmonary hypertension is also known to impair myocardial perfusion, despite presence of normal coronary vasculature. Whilst little is known about the maladaptation of the RV branches of the right coronary artery to RV remodelling and failure, several mechanisms have been proposed. Some degree of baseline hypoxaemia is frequently observed in CTEPH patients due to presence of ventilation-perfusion mismatch. Increasing wall tension described above increases myocardial oxygen demand whilst simultaneously reducing coronary circulation and therefore decreasing oxygen supply. Mismatch of coronary microcirculation and hypertrophied cardiac myocytes may lead to myocyte ischaemia and apoptosis. A combination of these factors is likely to further compound RV dysfunction and right heart failure in CTEPH [25].
Remodelling of the RV in CTEPH may also cause dilatation of the tricuspid annulus, leading to functional tricuspid regurgitation (TR). Ongoing TR may lead to dilatation of the right atrium, increase of RV preload and decreased RV stroke volume which further compounds right heart failure [27]. Given the functional nature of TR secondary to CTEPH, repair is rarely necessary during PEA because the reduction of PVR and postsurgical remodelling reduce regurgitation. Interaction between the right and left ventricle, characterised as ventricular interdependence (see Pathophysiology chapter), could explain the decrease in LV chamber size, compliance and contractility and overall stroke volume. Furthermore, RV dyssynchrony, secondary to RV remodelling and RV-LV diastolic interventricular delay, may contribute to impairment of LV filling and effective stroke volume [28, 29].
The progression from symptomatic PE to right heart failure in CTEPH follows a highly variable time course between patients. In addition to the above-described morphologic and haemodynamic processes, superimposed altered gene expression, maladaptive neurohormonal activation, oxidative stress and inflammatory responses have been implicated in the progression of heart failure and may explain individual variability [27].
9.3.3 Clinical Presentation and Diagnosis
The lack of specific clinical signs and symptoms complicates early diagnosis in CTEPH, with a multinational registry reporting a median time of 14.1 months between first onset of symptoms and diagnosis [30]. A ‘honeymoon period’ lasting between months and several years is often described after acute initiating embolic event and development of CTEPH symptoms. Patients typically present with progressive exertional dyspnoea, fatigue and, later in the disease course, signs of right heart failure including chest pain, peripheral oedema and syncope. Compared to idiopathic pulmonary hypertension, CTEPH is more likely to follow an episodic disease course and present more often with haemoptysis. Following a thorough history identifying above-described risk factors for VTE, coagulopathy and dysfibrinogenaemia, physical findings on examination may include a prominent S2 pulmonary component, a systolic murmur of secondary TR, left parasternal heave and/or signs of right heart failure [30]. CTEPH should be considered as a differential in any patient presented with otherwise unexplained pulmonary hypertension.
Echocardiography is widely employed as the initial diagnostic tool to indirectly evaluate pulmonary artery pressures and excludes cardiac causes of pulmonary hypertension. Despite lacking the specificity of other modalities, follow-up echocardiography in patients following acute PE with evidence for right heart failure or pulmonary hypertension is recommended by the European Society of Cardiology and European Respiratory Society [31]. Although there are ongoing developments in computed tomography pulmonary angiography (CTPA) and magnetic resonance pulmonary imaging (MRI), ventilation/perfusion (VQ) planar scans of at least 6 views remain the preferred initial diagnostic modality for CTEPH. Sensitivity in detecting CTEPH with VQ scan is reported >95 % compared to 51 % with CTPA in older studies, but CTPA technology has advanced since and is probably similar with experienced radiological reporting. A technically appropriate series of VQ scans will identify at least one, but often several segmental or larger mismatched perfusion defects in CTEPH, and where no perfusion defect is noted, CTEPH may be ruled out. In comparison, multidetector CT can detect other changes in CTEPH including organised emboli, dilated pulmonary arteries, filling defects, enlargement of right heart chambers, band and web formation and mosaic perfusion pattern of the lung parenchyma. Most CTEPH specialist centres correlate multiple imaging modalities to initially confirm the diagnosis of CTEPH and also assess operability [32, 33]. Right heart catheterisation (RHC) is essential for diagnosis of PH. RHC enables direct measurement of PA pressure and indirect calculation of PVR.
Using a combination of imaging and RHC measurements in a patient effectively anticoagulated >3 months, CTEPH is thus defined as the presence of the following combination of features [34]:
1.
Mean pulmonary artery pressure (mPAP) ≥25 mmHg
2.
Pulmonary capillary wedge pressure ≤15 mmHg
3.




Evidence of one or more mismatched subsegmental or larger perfusion defects on VQ scan, multidetector CTPA or conventional pulmonary angiography
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
