Chapter 57 Pulmonary Embolism
Epidemiology, Risk Factors, and Pathogenesis
Risk Factors
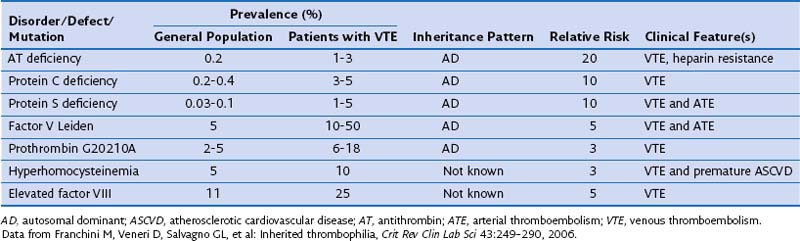
Inherited Thrombophilia
Inherited thrombophilias may result from qualitative or quantitative defects in coagulation factor inhibitors (antithrombin, protein C, protein S), increased levels or function of coagulation factors (activated protein C resistance, factor V Leiden mutation, prothrombin gene mutation, elevated factor VIII levels), hyperhomocysteinemia, defects in fibrolysis, or altered platelet function. Epidemiologic features of the common inherited thrombophilias are shown in Table 57-1.
Acquired Risk Factors
Acquired risk factors for VTE are far more prevalent than inherited thrombophilias. Box 57-1 lists common risk factors for acquired VTE.
Box 57-1
Acquired Risk Factors for Venous Thromboembolism
Clinical Features
The clinical consequences of PE range from incidental and clinically unimportant to circulatory collapse and sudden death. Equally challenging, the clinical signs and symptoms related to PE are diverse and nonspecific. Therefore, clinicians use a combination of history and examination findings in association with clinical prediction tools to determine appropriate diagnostic tests and the need for therapeutic interventions. Considerations in the differential diagnosis for acute PE are listed in Box 57-2.



