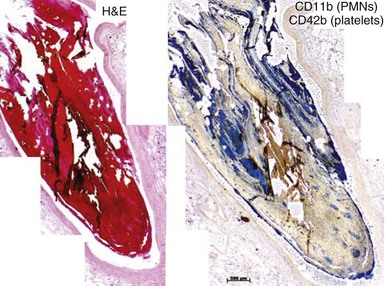
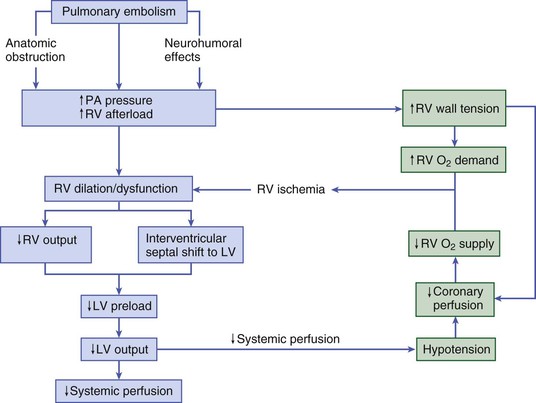
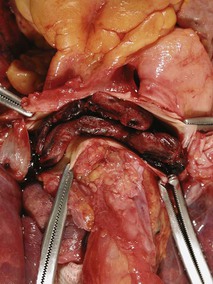
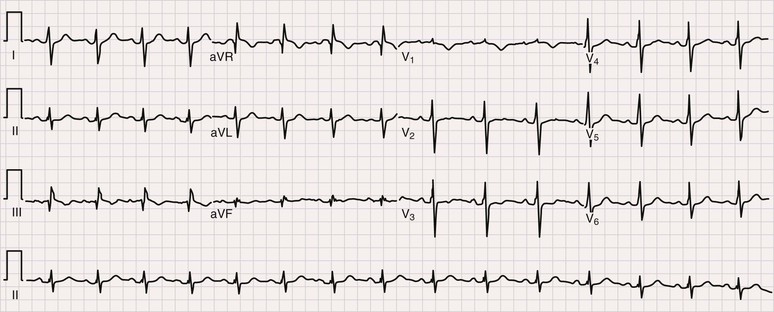
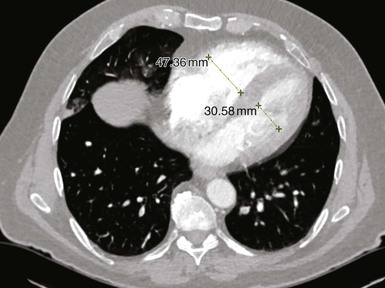
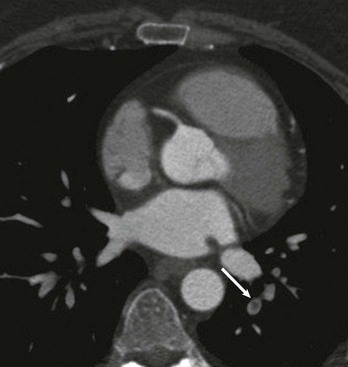
Samuel Z. Goldhaber Pulmonary embolism (PE) and deep vein thrombosis (DVT) together constitute one of the “big three” cardiovascular diseases, the other two being myocardial infarction (MI) and stroke. Venous thromboembolism (VTE) encompasses PE and DVT and causes more than 100,000 deaths annually in the United States. The in-hospital case-fatality rate for PE is approximately 7%. Most deaths in hospitalized patients with PE result from right-heart failure from the initial PE, or from recurrent PE developing despite adequate anticoagulation. The in-hospital case-fatality rate for patients who present with hemodynamic instability is approximately 30%, 10-fold higher than for patients who are hemodynamically stable.1 Hospital costs to manage acute PE are staggering—at Brigham and Women’s Hospital in Boston, for example, the actual expenses exceed $1 million per year.2 In a sample of U.S. acute care hospitals, total hospital charges per patient with PE increased in an 8-year period from $25,293 to $43,740.3 PE impairs quality of life4 and causes major long-term complications, including recurrent VTE, chronic thromboembolic pulmonary hypertension (CTEPH),5 and postthrombotic syndrome (also called chronic venous insufficiency) involving the legs.6 In 892 Dutch patients with PE, more than half suffered recurrent VTE, CTEPH, cancer, arterial cardiovascular events, or death from comorbid conditions within 4 years of follow-up.7 Among 1023 Australian patients hospitalized with PE and then followed over the long term after discharge, the cumulative mortality rate was 32% over 5 years, with 40% of the deaths attributed to cardiovascular causes. Postdischarge mortality was 2.5 times higher than in an age-matched and sex-matched population.8 PE and DVT increase in frequency with age but afflict children and teenagers9 as well as elderly persons. Recurrence after completion of a time-limited course of anticoagulation occurs often, especially when surgery, trauma, or estrogens do not precipitate the initial event. VTE exacts a psychological toll on patients, who wonder whether they will suffer a recurrent event and worry about the associated burden on their families and its implications including diminished quality of life and shortened life span. Advances in diagnostic, therapeutic, and preventive strategies, coupled with major leaps forward in the current understanding of pathophysiology, continue to emerge at an unprecedented rapid pace. Clinical and electronic decision tools lead to early detection and improves prevention strategies. Availability of novel oral anticoagulants such as rivaroxaban allows the management of PE and DVT without any parenteral anticoagulation for a majority of patients who suffer a VTE. With the recent recognition of critical role of the activated platelet in VTE pathogenesis, low-dose aspirin provides broader management options. For patients requiring advanced therapy, new invasive tools such as ultrasound-facilitated and catheter-assisted thrombolysis with low-dose tissue plasminogen activator therapy promise a lower rate of hemorrhagic complications than that associated with traditional, systemically administered thrombolysis. Intertwining risk factors and pathophysiology link VTE and atherothrombosis.10 VTE is now regarded as a pan-cardiovascular syndrome that includes coronary artery disease, peripheral artery disease, and cerebrovascular disease. Inflammation, hypercoagulability, and endothelial injury activate the pathophysiologic cascade leading to VTE. Venous thrombi contain fibrin, red blood cells, platelets, and neutrophils (Fig. 73-1). These thrombi flourish in an environment of stasis, low oxygen tension, oxidative stress, increased expression of proinflammatory gene products, and impaired endothelial cell regulatory capacity. Inflammation resulting from infection, transfusion, or erythropoiesis-stimulating factor11 activates a cascade of biochemical reactions in the vein endothelium that promotes thrombosis.12 Chronic kidney disease is associated with VTE,13 probably because impaired kidney function causes heightened states of oxidative stress and inflammation.14 PE can elicit a complex cardiopulmonary response that includes increased pulmonary vascular resistance due to vascular obstruction, neurohumoral agents, or pulmonary artery baroreceptors; impaired gas exchange caused by increased alveolar dead space from vascular obstruction and hypoxemia from alveolar hypoventilation and right-to-left shunting, as well as impaired carbon monoxide transfer caused by loss of gas exchange surface; alveolar hyperventilation caused by reflex stimulation of irritant receptors; increased airway resistance due to bronchoconstriction; and decreased pulmonary compliance due to lung edema, lung hemorrhage, and loss of surfactant. The extent of pulmonary vascular obstruction, the presence of underlying cardiopulmonary disease, and the neurohumoral response determine whether right ventricular dysfunction ensues. As obstruction increases, pulmonary artery pressure rises. Further increases in pulmonary vascular resistance and pulmonary hypertension result from secretion of vasoconstricting compounds such as serotonin, reflex pulmonary artery vasoconstriction, and hypoxemia. The overloaded right ventricle releases cardiac biomarkers such as pro–B type natriuretic peptide (pro-BNP), brain natriuretic peptide (BNP), and troponin, all of which portend an increased likelihood of adverse clinical outcomes. The sudden rise in pulmonary artery pressure abruptly increases right ventricular afterload, with consequent elevation of right ventricular wall tension followed by right ventricular dilation and dysfunction (Fig. 73-2). As the right ventricle dilates, the interventricular septum shifts toward the left, leading to underfilling and decreased left ventricular diastolic distensibility. With hampered filling of the left ventricle, systemic cardiac output and systolic arterial pressure both decline, impairing coronary perfusion and producing myocardial ischemia. Elevated right ventricular wall tension after massive PE reduces right coronary artery flow and increases right ventricular myocardial oxygen demand, causing ischemia. Perpetuation of this cycle can lead to right ventricular infarction, circulatory collapse, and death. Classification of acute PE (Table 73-1) can assist with prognostication and clinical management.20 Massive PE accounts for 5% to 10% of cases. Submassive PE is more common, occurring in approximately 20% to 25% of patients. Low-risk PE constitutes the majority of PE cases—approximately 70%. TABLE 73-1 Classification of Acute Pulmonary Embolism UFH = unfractionated heparin. Patients with massive PE are susceptible to cardiogenic shock and multisystem organ failure. Renal insufficiency, hepatic dysfunction, and altered mentation are common findings. Thrombosis is widespread, affecting at least half of the pulmonary arterial vasculature. Clot typically is present bilaterally, sometimes as a “saddle” PE. Dyspnea usually is the most prominent symptom; chest pain is unusual, transient cyanosis is common, and systemic arterial hypotension requiring pressor support occurs frequently. Excessive fluid boluses may worsen right-sided heart failure, rendering therapy more difficult. Patients with submassive PE present with moderate or severe right ventricular hypokinesis as well as elevations in troponin, pro-BNP, or BNP, but they maintain normal systemic arterial pressure. Usually, one third or more of the pulmonary artery vasculature is obstructed in these patients. Sudden onset of moderate pulmonary arterial hypertension (Fig. 73-3) and right ventricular enlargement is common. If patients have no previous history of cardiopulmonary disease, they may appear clinically well, but this initial impression often is misleading. They are at risk for recurrent PE, even with adequate anticoagulation. Most survive but may require escalation of therapy with pressor support or mechanical ventilation.21 Those patients designated as having low-risk PE exhibit no markers of an adverse prognosis. They present with normal systemic arterial pressure, no cardiac biomarker release, and normal right ventricular function. They often prove to have an anatomically small PE and appear clinically stable. Adequate anticoagulation results in an excellent clinical outcome. Pulmonary infarction is characterized by pleuritic chest pain that may be unremitting or may wax and wane. The pleurisy occasionally is accompanied by hemoptysis. The embolus usually lodges in the peripheral pulmonary arterial tree, near the pleura (Fig. 73-4). Tissue infarction usually occurs 3 to 7 days after embolism. Signs and symptoms often include fever, leukocytosis, elevated erythrocyte sedimentation rate, and radiologic evidence of infarction. Paradoxical embolism may manifest with a sudden stroke, which may be misdiagnosed as “cryptogenic.” The cause is a DVT that embolizes to the arterial system, usually through a patent foramen ovale. The DVT can be small and break away completely from a tiny leg vein, leaving no residual evidence of thrombosis that can be imaged on venous ultrasound examination.22 Sources of embolism other than thrombus are uncommon. They include fat, tumor, air, and amniotic fluid. Fat embolism most often occurs after blunt trauma complicated by long bone fractures. Air embolus can occur during placement or removal of a central venous catheter. Amniotic fluid embolism may be catastrophic and is characterized by respiratory failure, cardiogenic shock, and disseminated intravascular coagulation. Intravenous drug abusers sometimes self-inject hair, talc, and cotton as contaminants of the drug of abuse; these patients also are susceptible to septic PE, which can cause endocarditis of the tricuspid or pulmonic valve. Patients present with DVT symptoms approximately three times more frequently than with symptoms of PE. Leg DVT occurs approximately 10 times more often than upper-extremity DVT. The more proximal the thrombus is within the deep leg veins, the more likely it is to embolize and cause acute PE. When venous thrombi detach from their sites of formation, they travel through the venous system toward the vena cava. They pass through the right atrium and right ventricle and then enter the pulmonary arterial circulation. An extremely large embolus may lodge at the bifurcation of the pulmonary artery, forming a saddle embolus (Fig. 73-5). In many patients with large PEs, ultrasonographic evidence of DVT is lacking, probably because the clot has already embolized to the lungs. Upper-extremity DVT is an increasingly important clinical entity owing to more frequent placement of pacemakers and implantable cardioverter-defibrillators, as well as more frequent use of chronic indwelling catheters for chemotherapy and nutrition.23 The likelihood of upper-extremity DVT increases as the size of a peripherally inserted central catheter increases.24 A hospital initiative to use smaller-diameter catheters and to minimize the number of lumens can markedly reduce the frequency of catheter-associated DVT.25 Patients with upper-extremity DVT are at risk for PE, superior vena cava syndrome, and loss of vascular access.26 Dysfunction of the valves of the deep venous system often results from damage secondary to DVT. Obstruction of the deep veins may limit the outflow of blood, causing increased venous pressure with muscle contraction. Abnormal hemodynamics in the large veins of the leg are transmitted into the microcirculation, causing venous microangiopathy. Physical findings may include varicose veins, abnormal pigmentation of the medial malleolus, and skin ulceration. The economic impact of postthrombotic syndrome is high27 because of time lost from work and the expense of medical diagnosis and treatment (Fig. 73-6). Chronic venous disease is associated with a reduced quality of life as a consequence of pain, decreased physical function, and decreased mobility. Vascular compression stockings (below-knee, 30 to 40 mm Hg) are a mainstay of therapy, improving venous hemodynamics, reducing edema, and minimizing skin discoloration. By alleviating calf discomfort, these stockings improve quality of life.28 In a large population-based case-control study, superficial venous thrombosis was associated with a sixfold increased risk of DVT and a fourfold increased risk of PE.29 In a separate French cohort study of 600 patients with superficial venous thrombosis but no VTE at baseline, 10% developed VTE or experienced extension or recurrence of superficial venous thrombosis within 3 months.30 Short-term use of fondaparinux (2.5 mg once daily for 45 days) is the most well-validated therapy, based on a randomized trial in 3002 patients,31 although this approach might not be cost-effective.32 Shared risk factors for VTE and atherothrombosis33 include increasing age, obesity, cigarette smoking, and diabetes mellitus.34 Patients with newly diagnosed VTE have an increased long-term risk for development of MI or stroke.35 A meta-analysis of data for 63,552 patients with VTE and control subjects found that the relative risk for VTE was 2.3 for obesity, 1.5 for hypertension, 1.4 for diabetes mellitus, 1.2 for cigarette smoking, and 1.2 for hypercholesterolemia. High-density lipoprotein (HDL) cholesterol levels were lower in patients with VTE.36 Persistent stress,37 as well as depression and loneliness,38 also predisposes to VTE. In addition, seasonal variability has been documented for VTE: Its incidence increases in winter months.39 The obesity epidemic presents an increasing burden of risk for VTE. In a study of more than 1 million women with a mean age of 56 years in the United Kingdom, VTE risk increased with increasing body mass index (BMI). Women with a BMI 35 kg/m2 or greater, for example, were three to four times more likely to develop VTE than women with a BMI between 22 and 25 kg/m2.40 In a cohort study of almost 70,000 female nurses, physical inactivity, measured in hours of sitting each day, was associated with more than double the risk of PE.41 The U.S. National Hospital Discharge Survey of 14,721 PE patients found an increased likelihood of in-hospital death among patients 50 years of age or older, as well as in patients with concomitant cancer, pneumonia, or fracture.42 A Spanish VTE registry called RIETE (Registro Informatizado de Enfermedad TromboEmbólica) included 18,023 patients with PE. Immobilized patients had a more than twofold increased risk for fatal PE. Of RIETE patients dying from PE, 43% had a history of recent immobilization for 4 days or longer.43 The metabolic syndrome also is associated with VTE.44 Heart failure and chronic obstructive pulmonary disease (COPD) are potent risk factors for in-hospital death among patients with VTE. The overlap between venous and arterial thrombosis risk factors means that clinicians can counsel patients on steps to reduce VTE and coronary heart disease risk simultaneously. The incidence of VTE is approximately 1.5 cases per 1000 person-years, and DVT cases are approximately twice as numerous as PE cases. Incidence increases with age and is similar in men and women. Approximately half of the cases are idiopathic, occurring without antecedent trauma, surgery, immobilization, or cancer. VTE aggregates in families. In a Swedish registry of 45,362 patients with VTE, genetic rather than environmental factors explained most of the familial risk.45 Clinical predictors of fatal PE include black race,46 obesity, anatomically massive PE, neurologic disease, age older than 75 years, and cancer.47 Some risk factors for VTE, such as autoimmune disorders,48 are not readily modifiable (Table 73-2). As patients survive longer with cancer, the frequency of VTE is increasing, because these patients have a four- to sevenfold increased incidence of VTE49 (see Chapter 69). Cancer chemotherapy–associated VTE is a common problem.50 Increased VTE risk not only accompanies adenocarcinomas of the pancreas, stomach, lung, esophagus, prostate, and colon but also threatens patients with “liquid tumors” such as myeloproliferative disorders, lymphoma, and leukemia. In patients with a first VTE and without the diagnosis of cancer, the risk for detection of a subsequent cancer is 1% to 2% per year and is higher in patients with unprovoked VTE and in older patients, as assessed in 10-year increments.51 TABLE 73-2 Major Risk Factors for Venous Thromboembolism That Are Not Readily Modifiable Pregnancy, hormonal contraception, and postmenopausal hormonal therapy each contribute to increased risk. Use of progesterone-only birth control pills is not associated with increased VTE risk.52 Long-haul air travel is among the most frequently discussed acquired risk factors, although the associated risk of fatal PE is less than 1/1 million. When death occurs, however, it is dramatic and perceived as especially tragic because the victim often is an otherwise healthy young person. For each 2-hour increase in travel duration, there appears to be an 18% higher risk of VTE.53 Of 26,172 patients with VTE enrolled in RIETE, 2% had travel-related VTE. The most common risk factors were high BMI, previous VTE, hormone use, and thrombophilia.54 Hospitalized patients with conditions such as pneumonia, heart failure, and COPD are at high risk for development of VTE. The stasis and immobilization associated with postoperative venous thrombosis may paradoxically increase after hospital discharge, because with short hospital stays, patients may be too weak and debilitated to walk at home. Risk factors for VTE in the community include advancing age, cancer, previous VTE, venous insufficiency, pregnancy, trauma, frailty, and immobility. Of those suffering VTE in the Worcester Venous Thromboembolism Study, 23% had undergone surgery and 36% had been hospitalized within the preceding 3 months. Among those patients, fewer than half had received anticoagulant prophylaxis.55 More than half of VTE events occurred in subjects who were 65 years of age or older.56 Classically, the pathogenesis of PE has been dichotomized as caused by either inherited (primary) or acquired (secondary) risk factors. A combination of thrombophilia and acquired risk factors, however, usually precipitate thrombosis. The two most common identified genetic causes of thrombophilia are factor V Leiden57 and the prothrombin gene mutation (see Chapter 82). Normally, a specified amount of activated protein C (aPC) can be added to plasma to prolong the activated partial thromboplastin time (aPTT). Patients with “aPC resistance” exhibit blunted aPTT prolongation and are predisposed to the development of PE and DVT. The phenotype of aPC resistance is associated with a single-point mutation, designated factor V Leiden, in the factor V gene. Factor V Leiden triples the risk of VTE and is associated with recurrent pregnancy loss, probably as a consequence of placental vein thrombosis. Use of oral estrogen-containing contraceptives by patients with factor V Leiden increases the VTE risk by at least 10-fold. A single-point mutation in the 3′ untranslated region of the prothrombin gene (G-to-A transition at nucleotide position 20210) is associated with increased levels of prothrombin. The prothrombin gene mutation doubles the risk of VTE. The antiphospholipid syndrome, the most common acquired thrombophilia, can cause venous or arterial thrombosis, thrombocytopenia, recurrent fetal loss, or acute ischemic encephalopathy. Obtaining a family history remains the fastest and most cost-effective method of identifying a predisposition to venous thrombosis. Investigation with blood tests to detect known causes of hypercoagulability can be misleading. Consumption coagulopathy caused by venous thrombosis, for example, may be misdiagnosed as deficiency of antithrombin, protein C, or protein S. Heparin administration can depress antithrombin levels. Use of warfarin ordinarily causes a mild deficiency of protein C or protein S. Both oral contraceptives and pregnancy depress protein S levels. One of the greatest challenges in diagnosing PE is that it can masquerade as other illnesses, such as asthma, pneumonia, pleurisy, acute coronary syndrome, and congestive heart failure. PE often occurs concomitantly with other illnesses, thereby confounding the diagnostic workup. The most useful approach is a clinical assessment of likelihood, based on presenting symptoms and signs, in conjunction with judicious diagnostic testing. When PE is not among the most likely diagnoses, a normal plasma D-dimer enzyme-linked immunosorbent assay (ELISA) usually can rule out this condition.58 When PE is strongly suspected, a D-dimer ELISA need not be obtained; in most cases with high clinical suspicion, it is appropriate to proceed directly to chest computed tomography (CT) imaging.59 Symptoms and signs of PE are nonspecific. Hence clinical suspicion for PE is of paramount importance in guiding diagnostic testing.60 Dyspnea is the most frequent symptom, and tachypnea is the most frequent sign of PE (Table 73-3). Severe dyspnea, syncope, or cyanosis portends a major life-threatening PE, in which the clinical picture often is devoid of chest pain. Paradoxically, severe pleuritic pain often signifies that the embolism is small, not life-threatening, and located in the distal pulmonary arterial system, near the pleural lining. PE should be suspected in hypotensive patients when there is evidence of (1) venous thrombosis or predisposing VTE risk factors; (2) acute cor pulmonale (acute right ventricular failure), with features such as distended neck veins, right-sided S3 gallop, right ventricular heave, tachycardia, or tachypnea; especially if (3) there are echocardiographic findings of right ventricular dilation and hypokinesis or electrocardiographic evidence of acute cor pulmonale manifested by a new S1Q3T3 pattern (Fig. 73-7), new right bundle branch block, or right ventricular ischemia manifested by inferior T wave inversion or T wave inversion in leads V1 through V4. Clinical decision rules can stratify patients into groups with high clinical likelihood or non–high clinical likelihood of PE, using a set of seven bedside assessment questions known as the Wells criteria (Table 73-4). TABLE 73-4 Classic Wells Criteria to Assess Clinical Likelihood of Pulmonary Embolism * >4 score points = high probability; ≤4 score points = non–high probability. The differential diagnosis of PE is broad in scope, covering a wide spectrum from life-threatening conditions such as acute MI to anxiety states (Table 73-5). Concomitant PE and other illnesses should be taken into account—for example, if pneumonia or heart failure does not respond to appropriate therapy, the possibility of coexisting PE should be considered. Idiopathic pulmonary hypertension may manifest with sudden exacerbations that mimic acute PE. The plasma D-dimer assay is a blood-screening test that relies on the following principle: Most patients with PE have ongoing endogenous fibrinolysis that is not effective enough to prevent PE but breaks down some of the fibrin clot to D-dimers. Although elevated plasma concentrations of D-dimers are sensitive for the presence of PE, they are not specific. Levels are elevated for at least 1 week postoperatively and also are abnormally high in patients with MI, sepsis, cancer, or almost any other systemic illness. The plasma D-dimer assay therefore is ideally suited for screening outpatients or emergency department patients who have suspected PE but no coexisting acute systemic illness. This test generally is not useful for screening acutely ill hospitalized inpatients because their D-dimer levels usually are elevated.60 The electrocardiogram (ECG) helps exclude acute MI and acute pericarditis. This test may help lead the clinician toward the diagnosis of PE among patients with electrocardiographic manifestations of right-sided heart strain, which is an ominous prognostic finding.61 Right-sided heart strain is not specific, however, and may be observed in patients with asthma or idiopathic pulmonary hypertension. In patients with massive PE, the ECG may exhibit sinus tachycardia, slight ST-segment and T wave abnormalities, or even an entirely normal appearance. A near-normal radiographic appearance in the setting of severe respiratory compromise is highly suggestive of massive PE. Major chest radiographic abnormalities are uncommon. Focal oligemia (Westermark sign) indicates massive central embolic occlusion. A peripheral wedge-shaped density above the diaphragm (Hampton hump) usually indicates pulmonary infarction (Fig. 73-4). A subtle abnormality suggestive of PE is enlargement of the descending right pulmonary artery. The chest radiograph also can help identify patients with diseases that mimic PE, such as lobar pneumonia and pneumothorax, but patients with these illnesses also can have concomitant PE. Pulmonary radionuclide perfusion scintigraphy (lung scanning) uses radiolabeled aggregates of albumin or microspheres that lodge in the pulmonary microvasculature. Patients with large PE often have multiple perfusion defects. If ventilation scanning is performed on a patient with PE but no intrinsic lung disease, a normal ventilation study result is expected, yielding ventilation-perfusion mismatch; the lung scan is then interpreted as indicating a high probability of PE. However, many patients with low-probability scans but with clinical findings strongly suggestive of PE do, in fact, have PE proven by invasive pulmonary angiography. Thus clinical probability assessment before obtaining a lung scan helps in correct interpretation of the scan results. Most lung scans are nondiagnostic. An unequivocal normal or high-probability scan is the exception, not the rule. Interobserver variability is common, even among experts. Three principal indications for obtaining a lung scan are renal insufficiency, anaphylaxis occurring in reaction to intravenous contrast agent that cannot be suppressed with high-dose corticosteroids, and pregnancy (lower radiation exposure to the fetus). Chest CT has supplanted pulmonary radionuclide perfusion scintigraphy as the initial imaging test in most patients with suspected PE, allowing ready visualization of massive PE and confirmation of surgical or catheter accessibility to the centrally located thrombus. Multidetector-row CT scanners can rapidly image the entire chest with submillimeter resolution. Three-dimensional images can be reconstructed, and color can be added electronically to enhance details of thrombus localization. The latest generation of scanners can image thrombus in sixth-order vessels. These thrombi are so tiny that their clinical significance is uncertain (Fig. 73-8). The chest CT scan also can detect other pulmonary diseases that manifest in conjunction with PE or explain a clinical presentation that mimics PE. These diseases include pneumonia, atelectasis, pneumothorax, and pleural effusion, which may not be well visualized on the chest radiograph. A chest CT scan sometimes detects an incidental but critical finding, such as a small lung carcinoma. Routine use of CT leg venography increases radiation exposure, and the findings rarely change clinical management. For patients with PE, the CT scan serves as a prognostic and diagnostic test. It shows a four-chamber view of the heart and images the pulmonary arteries. Careful evaluation of the CT scan can detect signs of right ventricular dysfunction by analyzing (1) right ventricular–to–left ventricular diameter ratio (Fig. 73-9), (2) right ventricular–to–left ventricular volume ratio, (3) interventricular septal bowing, and (4) reflux of contrast medium into the inferior vena cava.62
Pulmonary Embolism
State-of-the-Art
Molecular Pathophysiology
Cardiopulmonary Dynamics
Classification of Pulmonary Embolism
CATEGORY (FREQUENCY)
PRESENTATION
THERAPY
Massive PE (5%-10%)
Systolic blood pressure < 90 mm Hg or poor tissue perfusion or multisystem organ failure plus extensive thrombosis, such as “saddle” PE or right or left main pulmonary artery thrombus
Anticoagulation (usually starting with intravenous UFH), plus consider advanced therapy: systemic thrombolysis, pharmacomechanical catheter-directed therapy, surgical embolectomy, or inferior vena cava (IVC) filter
Submassive PE (20%-25%)
Hemodynamically stable but moderate or severe right ventricular dysfunction or enlargement, coupled with biomarker elevation indicative of right ventricular microinfarction and/or right ventricular pressure overload
Anticoagulation usually with intravenous UFH until decision made regarding implementation of advanced therapy; controversy centers on this group. For systemic thrombosis, reducing the rate of cardiovascular collapse and death must be balanced against the increased rate of hemorrhagic stroke.
For patients at low bleeding risk with severe right ventricular dysfunction, consider same interventions as for massive PE
Small to moderate PE (70%)
Normal hemodynamics and normal right ventricular size and function
Anticoagulation with parenteral therapy as a bridge to warfarin or, alternatively, with oral rivaroxaban regimen as monotherapy
Massive Pulmonary Embolism
Submassive Pulmonary Embolism
Low-Risk Pulmonary Embolism
Pulmonary Infarction
Paradoxical Embolism
Nonthrombotic Pulmonary Embolism
Classification of Deep Vein Thrombosis
Lower-Extremity Deep Vein Thrombosis and the Relationship Between Deep Vein Thrombosis and Pulmonary Embolism
Upper-Extremity Deep Vein Thrombosis
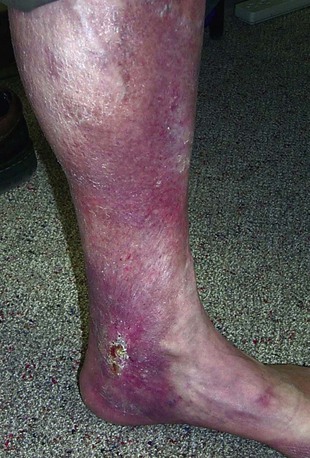
Postthrombotic Syndrome and Chronic Venous Insufficiency
Superficial Venous Thrombosis
Epidemiology
General Considerations
Clinical Risk Factors

Hypercoagulable States
Diagnosis
Clinical Presentation
CRITERION
SCORING*
DVT symptoms or signs
3
An alternative diagnosis is less likely than PE
3
Heart rate > 100 beats/min
1.5
Immobilization or surgery within 4 weeks
1.5
Previous DVT or PE
1.5
Hemoptysis
1
Cancer treated within 6 months or metastatic
1
Differential Diagnosis
Nonimaging Diagnostic Methods
Plasma D-Dimer Assay
Electrocardiogram
Imaging Methods
Chest Radiography
Lung Scanning
Chest Computed Tomography

Pulmonary Embolism
73