1. Pulmonary arterial hypertension (PAH)
1. Idiopathic PAH
2. Heritable PAH
1.2.1 BMPR2
1.2.2 ALK-1, ENG, SMAD9, CAV1, KCNK3
1.2.3 Unknown
1.3 Drug and toxin induced
1.4 Associated with
1.4.1 Connective tissue disease
1.4.2 HIV infection
1.4.3 Portal hypertension
1.4.4 Congenital heart diseases
1.4.5 Schistosomiasis
1′ Pulmonary venoocclusive disease and/or pulmonary capillary hemangiomatosis
1″ Persistent pulmonary hypertension of the newborn (PPHN)
2. Pulmonary hypertension due to left heart disease
1. Left ventricular systolic dysfunction
2. Left ventricular diastolic dysfunction
3. Valvular disease
4. Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies
3. Pulmonary hypertension due to lung diseases and/or hypoxia
1. Chronic obstructive pulmonary disease
2. Interstitial lung disease
3. Other pulmonary diseases with mixed restrictive and obstructive pattern
4. Sleep-disordered breathing
5. Alveolar hypoventilation disorders
6. Chronic exposure to high altitude
7. Developmental lung diseases
4. Chronic thromboembolic pulmonary hypertension (CTEPH)
5. Pulmonary hypertension with unclear multifactorial mechanisms
1. Hematologic disorders: chronic hemolytic anemia, myeloproliferative disorders, splenectomy
2. Systemic disorders: sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis
3. Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders
4. Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure, segmental PH
22.3 Pathology
22.3.1 The Pulmonary Vasculature
PAH predominantly affects the small resistance pulmonary arteries, characterized by intimal hyperplasia, medial hypertrophy, adventitial proliferation, in situ thrombosis, and inflammation (Fig. 22.1) [2]. Plexiform arteriopathy, which refers to capillary-like, angioproliferative vascular channels within the lumina of small muscular arteries, is the pathognomonic histopathological lesion of PAH [2]. Plexiform lesions often appear at branch points, frequently have fibrin thrombi within the lumen, and have varying channel diameter, giving them a disordered appearance.
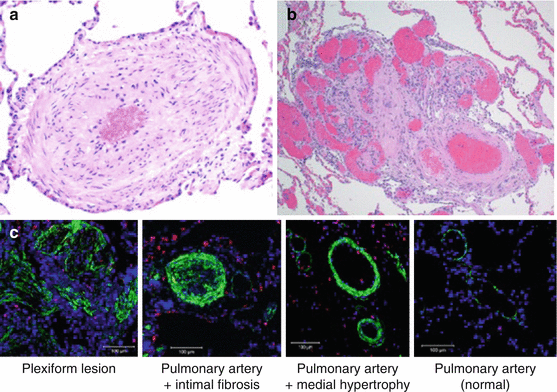
Fig. 22.1
Histopathological changes of the pulmonary vasculature in pulmonary arterial hypertension. (a) A small branch of pulmonary artery with severe medial hypertrophy and mild intimal fibrosis causing severe luminal obstruction (hematoxylin-eosin stain, original magnification ×20). (b) A characteristic plexiform lesion surrounded by dilated, thin-walled vessels (hematoxylin-eosin stain, original magnification ×4). (c) Immunofluorescent staining for proliferating cell nuclear antigen (PNCA) (red) indicative of ongoing vascular proliferation. The bright staining (green) is smooth muscle actin. The staining of the pulmonary vasculature from another patient who died without pulmonary vascular disease is shown for comparison as a normal control. Staining for PNCA is absent in the normal control. PAH pulmonary arterial hypertension (Reproduced with permission from the American College of Chest Physicians [2])
22.3.2 The Right Ventricle (RV)
The chronic elevation in RV afterload due to increased PVR induces right ventricular hypertrophy (RVH), which can be either adaptive or maladaptive (Fig. 22.2). Adaptive RVH, characterized by concentric hypertrophy with minimal eccentric dilatation and fibrosis, maintains normal ejection fraction, cardiac output, and filling pressures [2]. However, in contrast, maladaptive RVH illustrates eccentric dilatation, increased fibrosis, and capillary rarefaction with reduction in ejection fraction and cardiac output and elevation in filling pressures [2, 3]. At a metabolic level, maladaptive RVH is characterized by increased aerobic glycolysis and glutaminolysis [4]. In addition, maladaptive RVH is associated with increased sympathetic activation and downregulation of α, β, and dopaminergic receptors in the RV myocytes [5]. Some patients, especially those with congenital heart disease associated PAH, remain stable with adaptive RVH for a prolonged period. However, in contrast, certain PAH patients, specifically those with scleroderma-associated PAH, develop maladaptive RVH relatively early, leading to RV failure and death [6]. The mechanisms that switch the adaptive, compensatory hypertrophy of the RV to maladaptive RV dilatation and ultimately RV failure are unclear and are under investigation [5, 7]. Long-term outcomes in PAH are largely determined by the response of the RV to the increased afterload [8].
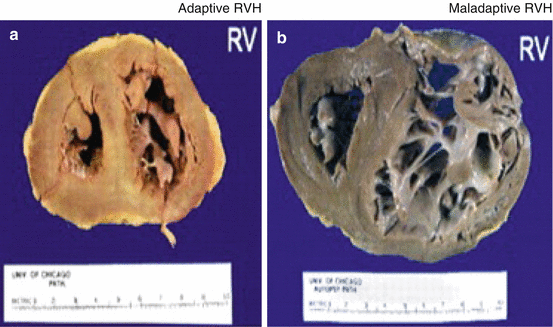
Fig. 22.2
Adaptive vs. maladaptive right ventricular hypertrophy in pulmonary arterial hypertension. (a) Adaptive right ventricular hypertrophy (RVH) characterized by concentric hypertrophy and minimal dilatation. (b) Maladaptive right ventricular hypertrophy characterized by eccentric dilatation. RV right ventricle, RVH right ventricular hypertrophy (Reproduced with permission from the American College of Chest Physicians [2])
22.4 Pathogenesis
The pathogenesis of PAH likely involves multiple pathways rather than a single mechanism [1]. First, there is endothelial dysfunction characterized by an imbalance of vasoactive and vasodilator substances in the small pulmonary arteries. There is increased production of thromboxane and decreased synthesis of prostacyclin. Thromboxane is a potent vasoconstrictor and activates proliferation of platelets, whereas prostacyclin is a potent vasodilator, inhibits smooth muscle proliferation, and has antiplatelet properties [1]. In addition, there is an increased level of endothelin, which is a strong vasoconstrictor and stimulates pulmonary artery smooth muscle proliferation [1]. Furthermore, there is a decreased level of nitric oxide, which is a potent vasodilator, inhibits smooth muscle proliferation, and inhibits platelet activation. Other vasoactive substances that have been implicated in the pathogenesis of PAH include serotonin and vasoactive intestinal polypeptide.
Second, there are several changes in the pulmonary artery smooth muscle cells that favor increased proliferation and decreased apoptosis including inappropriate activation of transcription factors hypoxia-inducible factor (HIF)-1 alpha and nuclear factor of activated T cells (NFAT), decreased expression of voltage-gated potassium channels (e.g., Kv1.5 and Kv2.1), de novo expression of the antiapoptotic protein survivin, and increased expression of transient receptor potential channels (TRPC) leading to calcium overload. Third, there increased activation of adventitial metalloproteinases, leading to adventitial remodeling. Finally, there are inflammatory infiltrates and activation of proinflammatory cytokines, suggesting that inflammation may play a role in the pathogenesis of PAH [9].
PAH can also be inherited. Approximately, 10 % of patients have hereditable PAH. Mutations in three genes involved in the transforming growth factor beta (TGF-ß) pathway, bone morphogenetic protein receptor (BMPR2), activin-like kinase, and endoglin have been identified in patients with hereditable PAH [10]. BMPR2 regulates vascular smooth muscle cell growth by activating the intracellular pathways of SMAD and LIM kinase. Loss of function mutation of the BMPR2 gene leads to decreased activation of SMAD, ultimately leading to increased proliferation of pulmonary artery smooth muscle cells. Recently, mutations in caveolin-1 and KCNK3 genes have also been identified in patients with hereditable PAH without mutations in genes encoding BMPR2 or other TGF-ß superfamily members [10]. Caveolin-1 is a membrane protein that forms caveolae, which are flask-shaped invaginations of the plasma membrane. Caveolae regulates membrane trafficking, cell signaling, cholesterol homeostasis, and mechanotransduction [10]. KCNK3 gene encodes a two-pore potassium channel expressed in pulmonary artery smooth muscle cells. This potassium channel modulates resting membrane potential, pulmonary vascular tone, and hypoxic pulmonary vasoconstriction [10].
22.5 Epidemiology
The landmark NIH registry initiated in 1981 collected data prospectively from 32 centers in the United States between 1981 and 1985 [11]. One hundred eighty seven patients with idiopathic PAH, hereditable PAH, or anorexigen-associated PAH were included in this registry. The mean age at the time of presentation was 36 ± 15 years, and female to male ratio was 1.7:1. The predominant race was white (85.4 %) followed by African Americans (12.3 %) and Hispanics (2.3 %). The mean time interval between onset of symptoms and diagnosis was 2 years.
Since there has been a considerable change in the understanding of the pathogenesis and classification of PAH in the last three decades, several contemporary PAH registries were developed to advance our knowledge on PAH epidemiology. Data from these contemporary PAH registries suggest that the epidemiology of PAH has changed significantly in the current era [12]. PAH continues to remain as an orphan disease. The incidence and prevalence of PAH vary between 2–7.6 cases per million population and 10.6–26 cases per million population, respectively. In the contemporary PAH registries, nearly half of the patients had idiopathic or hereditable PAH, and the rest had associated PAH. The most common etiology of associated PAH is connective tissue disease followed by congenital heart disease [13–15]. Scleroderma is the most common connective tissue disease associated with PAH. The mean age of the patients with idiopathic or hereditable PAH enrolled in the contemporary registries is significantly higher when compared to the NIH registry (45–65 years vs. 36 years). The female to male ratio continues to remain high in the contemporary PAH registries, especially in the US-based registries [12, 16]. In the REVEAL registry, 72.8 % were Caucasians, 12.2 % were African Americans, 8.9 % were Hispanics, 3.3 % were Asians or Pacific Islanders, and 2.8 were others or unknown [15].
Unfortunately, despite increasing awareness of PAH, there is still a significant delay between the onset of symptoms and the diagnosis of PAH. The mean interval between onset of symptoms and diagnosis of PAH in the contemporary registries ranged from 18 to 32 months (vs. 2 years in the NIH registry) [17, 18]. In the REVEAL registry, 20 % of patients had symptoms >2 years before diagnosis [18]. Unlike the NIH registry, patients in the contemporary registries have several comorbidities that include systemic hypertension, diabetes, coronary artery disease, and the metabolic syndrome.
22.6 Clinical Manifestations
Dyspnea at rest or with exertion is the most common presenting symptom of PAH. Other symptoms include fatigue, chest pain or pressure with exertion, lightheadedness, dizziness, syncope, and fluid retention. Typical physical exam findings of PAH include elevated jugular venous distension with a prominent “A” wave; a palpable right ventricular heave; a loud pulmonic component of the second heart sound; a right-sided S4; a holosystolic murmur secondary to tricuspid regurgitation and an early diastolic murmur, due to pulmonary regurgitation; and a right-sided S4. The presence of a prominent “V” wave in the jugular vein distension, a right-sided S3 gallop rhythm, hepatomegaly, ascites, and lower extremity edema may suggest underlying right heart failure.
22.7 Diagnostic Evaluation
The classic chest x-ray findings of PAH are prominent central pulmonary artery, decreased peripheral pulmonary vascular markings (vascular pruning), and reduced retrosternal space on a lateral projection suggestive of RVH [9]. The typical EKG findings of PAH include right atrial enlargement, RVH with strain pattern, right axis deviation of the QRS complex, and QT interval prolongation. Both chest x-ray and EKG findings are neither sensitive nor specific for the diagnosis of PAH [9].
Transthoracic echocardiogram is the initial screening test of choice in patients suspected of having PAH based on history, physical examination, chest x-ray, and electrocardiogram or in patients with substrates, which increase the chances of pulmonary vascular disease (connective tissue disease, portal hypertension, HIV infection, etc.) [9]. A Doppler-estimated systolic PAP >40 mmHg, the presence of right atrial enlargement, RV enlargement, and flattened interventricular septum (D-shaped) should prompt further evaluation for PAH. Echocardiogram also helps to exclude other common cardiovascular causes of unexplained dyspnea including left ventricular systolic and diastolic dysfunction and valvular heart disease, which are commonly associated with PAH (WHO group 2 PAH). In patients suspected to have underlying congenital heart disease, a bubble study should be performed to identify intracardiac shunt. Transthoracic echocardiogram is a useful screening test; however, Doppler estimates of PAP can be inaccurate and should not be relied upon solely to make a definitive diagnosis of PAH. Doppler-based methods have been shown to either overestimate or underestimate systolic PAP by 10 mmHg in nearly half of the patients [19, 20].
Echocardiographic findings suggestive of PAH should prompt further thorough systematic evaluation to exclude other WHO categories of PH and to identify the presence of associated causes of PAH including congenital heart disease, connective tissue disease, portal hypertension, HIV infection, and exposure to anorexigen use. Idiopathic PAH should be a diagnosis of exclusion.
Pulmonary function test should be performed to exclude obstructive and restrictive lung disease, which are commonly associated with PH (WHO group 3). Although not sensitive or specific, a very low diffusion capacity for carbon monoxide would suggest PAH [21]. Overnight, polysomnography is recommended to exclude sleep-disordered breathing, which can lead to PH [22]. V/Q scan is the diagnostic test of choice for excluding chronic pulmonary thromboembolic disease (WHO group 4) [23]. A low or intermediate probability scan excludes chronic thromboembolic disease, whereas a high probability scan should prompt invasive pulmonary angiogram or CT angiogram of the chest for confirmatory diagnosis. It is important to exclude chronic thromboembolic PH since it can be potentially cured by surgical pulmonary thromboendarterectomy in suitable candidates. Although chest CT angiogram is highly sensitive and specific for diagnosis proximal pulmonary embolism, it is less sensitive for excluding distal chronic thromboembolic disease compared to V/Q scan. Evaluation should also include testing to exclude common conditions associated with PAH. Patients should be tested for human immunodeficiency virus (HIV) infection, antinuclear antibody to exclude connective tissue disease, and liver function tests to rule out portal hypertension.
Right heart catheterization is the gold standard test for diagnosing PAH [9]. It helps to confirm the diagnosis, to assess the severity, and to test for acute vasodilator response. PAH is defined as mean PAP greater than 25 mmHg at rest with a PCWP ≤15 mmHg and a PVR >3 Wood units. Accurate measurement of PCWP is crucial for the distinction of PAH (WHO group 1) from PH due to left heart disease (WHO group 2). A PCWP ≤15 mmHg excludes left heart disease. Left ventricular end-diastolic pressure should be measured if the accuracy of wedge pressure measurement is questionable [24]. Cardiac output measured by thermodilution method correlates well with the Fick method even in the presence of severe tricuspid regurgitation and low cardiac output in patients with PAH [25]. Acute vasodilator challenge with inhaled nitric oxide, inhaled epoprostenol, or intravenous adenosine should be performed after confirming the diagnosis of PAH, especially in patients with idiopathic PAH. A positive vasodilator response is defined as a fall in the mean PAP by 10 mmHg with an absolute value less than 40 mmHg and no change or increase in cardiac output.
Cardiac magnetic resonance imaging (CMRI) is the gold standard imaging modality for accurate and reproducible measurement of RV volumes, mass, and other markers of RV function. Patients with PAH typically have late gadolinium enhancement in the interventricular septum at the RV free-wall insertion sites, which has been associated with PAH severity and has been shown to be a univariate predictor of mortality [26].
22.8 Treatment
Management of patients with PAH includes general supportive measures and PAH-specific vasodilator therapy. The goals of therapy are to decrease symptoms, improve hemodynamics, reduce hospitalizations, and ultimately prolong survival.
22.8.1 General Supportive Measures
The general supportive treatment measures for PAH include supplemental nasal oxygen, diuretics as needed for right heart failure, and long-term anticoagulation with warfarin. A systematic review and meta-analysis based on two prospective and seven retrospective studies regarding warfarin in PAH suggest a survival benefit [27]. Long-term anticoagulation is recommended mainly for patients with PAH, especially idiopathic PAH [9]. The target INR is 1.5–2.5. There is limited data to support the role of digoxin for right ventricular failure from PAH. It is used in the presence of atrial arrhythmias.
22.8.2 Calcium Channel Blockers
Calcium channel blockers are indicated in patients with idiopathic PAH who have a positive response during acute vasodilator testing at the time of diagnostic right heart catheterization. Compared to nonresponders, in prospective nonrandomized studies, patients with a positive vasodilator response had a sustained and significant reduction in mean PAP on long-term oral calcium channel blocker therapy and a better prognosis [28, 29]. Only 5–10 % of patients with idiopathic PAH have a positive vasodilator response [13]. Patients treated with calcium channel blockers should be closely monitored for adequate response. The prevalence of responders in patients with associated PAH is very uncommon.
22.8.3 PAH-Specific Vasodilator Therapies
Four classes of pulmonary vasodilator therapy have been approved for the treatment of PAH: phosphodiesterase-5A-inhibitors, endothelin receptor antagonists, soluble guanylyl cyclase stimulators, and prostanoids. These approved drugs act primarily on the three primary pathways implicated in the pathogenesis of PAH: the prostanoid pathway, the endothelin pathway, and the nitric oxide pathway.
22.8.3.1 Phosphodiesterase-5A-inhibitors (PDE5A-Inhibitors)
PDE5A-inhibitors increase cyclic guanine monophosphate (cGMP), by inhibiting its hydrolysis by the enzyme PDE-5, which has vasodilators, antiproliferative, and proapoptotic effects. Currently, there are two oral PDE5A inhibitors, sildenafil and tadalafil approved for the treatment of PAH in patients with WHO functional class II or III symptoms. Sildenafil was approved based on the SUPER-1 trial that randomized sildenafil 20, 40, and 80 mg three times daily vs. placebo for 12 weeks in 278 patients with PAH [30]. Sildenafil increased 6-min walk distance, improved functional class, reduced mean PAP, increased cardiac output, and decreased PVR. A long-term extension study reported sustained improvement in 6-min walk distance at 1 year in 229 PAH patients treated with sildenafil 80 mg three times daily [30]. Tadalafil was approved based on the PHIRST trial that randomized 409 patients with PAH to placebo vs. 2.5, 10, 20, and 40 mg once daily [31]. In this trial, tadalafil increased 6-min walk distance, improved time to clinical worsening, incidence of clinical worsening, and health-related quality of life. PHIRST 2, an uncontrolled 52-week extension study, showed sustained improvement in 6-min walk distance with tadalafil [32].
22.8.3.2 Endothelin Receptor Antagonists (ERA)
Bosentan, ambrisentan, and macitentan are the three ERAs that are currently approved as a first-line therapy for PAH. Bosentan is a dual endothelin receptor antagonist that blocks both the endothelin A and B receptors. BREATH 2 randomized 213 patients with PAH either to receive placebo or bosentan 62.5 mg twice daily for 4 weeks followed by 125 mg twice daily or 250 mg twice daily for a minimum of 12 weeks. Bosentan increased 6-min walk distance, improved functional class, and increased time to clinical worsening [33]. Ambrisentan is a specific endothelin A receptor antagonists approved for PAH patients with WHO functional class II or III symptoms based on the ARIES 1 and ARIES 2 clinical trials [34]. ARIES 1 randomized 202 patients with PAH to placebo vs. 5 or 10 mg once daily dose of ambrisentan. ARIES 2 randomized 102 patients with PAH to placebo vs. 2.5 or 5 mg once daily dose of ambrisentan. Six-minute walk distance improved across all doses of ambrisentan with sustained benefits in a 48-week long-term extension study. Macitentan is a dual endothelin receptor antagonist that was recently approved for use in PAH patients with WHO functional class II or III symptoms. Macitentan significantly reduced morbidity and mortality in an event-driven long-term, placebo-controlled clinical trial [35].
22.8.4 Soluble Guanylyl Cyclase Stimulators
Riociguat, a soluble guanylyl cyclase stimulator, increases cGMP by directly stimulating the enzyme soluble guanylyl cyclase independent of nitric oxide. Increased cGMP causes vasodilatation and reduced inflammation and thrombosis. In the PATENT trial, compared to placebo, riociguat improved 6-min walk distance, increased time to clinical worsening, and decreased both PVR and NT-proBNP levels [36]. Riociguat is approved as a first-line therapy in PAH patients with WHO functional II or III symptoms.
22.8.5 Parenteral Prostanoids
Parental prostacyclin therapy remains the first-line treatment for PAH patients with WHO functional classification IV symptoms due to right ventricular failure. Epoprostenol improved exercise capacity, hemodynamics, quality of life, and delayed lung transplantation in PAH patients with functional class III/IV symptoms A [37]. Long-term observational studies have reported improved survival with epoprostenol treatment compared to historical controls and risk model predicted survival [38, 39]. Epoprostenol has a half-life of only 3–5 min, as it is very unstable at room temperature. Hence, it requires continuous intravenous infusion through central venous catheter, portable pump, and ice packs to maintain the stability. In general, only designated PAH centers with experienced physicians administer epoprostenol because of the complexity involved. Treprostinil is a tricyclic benzene prostacyclin analog. Its pharmacological properties are similar to epoprostenol but it has a longer half-life, and it is stable in room temperature, allowing it to be administered either by subcutaneous or intravenous routes and alleviating the need for ice packs. Treprostinil improved exercise capacity, hemodynamics, and long-term survival compared to survival predicted by the NIH equation [40, 41].
22.8.6 Inhaled Prostanoids
Iloprost is an inhalational prostacyclin approved for treatment of patients with PAH and WHO functional class III symptoms. Iloprost improved exercise capacity either as monotherapy or add on therapy to patients on background [42]. It has a much shorter half-life requiring six to nine times daily dosing. Treprostinil can also be administered by inhalation. In the TRIUMP study, inhaled treprostinil added on to patients with persistent symptoms on background sildenafil or bosentan therapy improved exercise capacity and quality of life [43]. Unlike iloprost, inhaled treprostinil is administered four times daily, which increases patient compliance.
22.8.7 Oral Prostanoids
Treprostinil diolamine, a sustained release oral formulation of treprostinil, is approved in the United States as a first-line therapy in PAH patients with WHO functional II and III symptoms. In the Freedom C trial, oral treprostinil improved exercise capacity in patients not on background therapy [44]. In the subsequent Freedom C2 trial, oral treprostinil added to background ERA and PDE5A-inhibtior therapy did not result in a statistically significant improvement in exercise capacity [45]. Beraprost is an oral prostanoid approved for use only in Japan. It is currently under investigation in the United States.
22.8.8 Combination Therapy
There is limited data on the role of combination therapy in PAH. In the PACES trial, addition of oral sildenafil in patients who were having persistent or worsening symptoms on background intravenous epoprostenol therapy improved 6-min walk distance, hemodynamics, time to clinical worsening, and quality of life [46]. In the TRIUMP study, addition of inhaled treprostinil in symptomatic patients on background bosentan or sildenafil improved 6-min walk distance and quality of life [43]. There are several ongoing clinical trials evaluating the role of various combination therapies including add-on therapy and upfront combination therapy. At present, upfront combination therapy at the time of diagnosis is still investigational.
22.8.9 Lung Transplantation
Lung transplantation is a potential therapeutic option for patients with PAH who do not respond to pulmonary vasodilator therapy. There is no consensus on single lung transplant vs. double lung transplant. Donor lung reperfusion injury has been reported in patients with PAH patients who undergo single lung transplants. Patients with severe right ventricular failure may need combined heart-lung transplant [9].
22.8.10 Monitoring and Goal-Oriented Therapy
PAH patients should be followed every 3–6 months. During follow-up, patients are characterized into three categories based on the clinical symptoms and signs, noninvasive evaluation, and invasive evaluation: stable and satisfactory, stable but not satisfactory, and unstable and deteriorating [47].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


