18 Pseudoneoplastic Lesions of the Lungs and Pleural Surfaces
There is a limited group of pulmonary lesions that one can classify as pseudoneoplastic, but their conditions comprise a significant aggregation in absolute numbers. Some are categorized as malformative or reactive, including pulmonary hamartomas; selected inflammatory pseudotumors (“plasma cell granulomas”); tumefactive lymphoid hyperplasias; inflammatory or reparative conditions simulating carcinomas; unusual granulomatous reactions; tumefactive pleural plaques; and florid examples of mesothelial hyperplasia. Other clinical “pseudotumors” such as amyloidoma and rounded atelectasis1 are confused with neoplasms only by nonpathologists and are not included for discussion here. On the other hand, the variant of inflammatory pseudotumor now known as inflammatory myofibroblastic tumor demonstrates clonal characteristics and can rightly be regarded as a true neoplasm.2,3 Accordingly, it likewise has been omitted from this chapter.
Pulmonary Hamartoma
The term hamartoma is intended to denote tumefactive malformations that exhibit an architecturally abnormal relationship between tissue components that are appropriate to the organ site in which they arise.4 Other terms for these lesions in the lung include benign mesenchymoma, fibroma, chondroma, fibrochondrolipoma, fibrolipomyochondroma, hamartoma-chondroma, cartilage-containing tumor of the lung, adenochondroma, lipochondroadenoma, adenofibrolipochondromyxoma, and mixed tumor.5 Although in the past, chondroma has been used interchangeably with the lesion referred to here as hamartoma, they constitute distinct lesions and are discussed in Chapter 19.
Pulmonary hamartomas (PHs) have been encountered in approximately 0.25% of autopsies.6 They can arise both centrally and peripherally, and the latter are more common in men than women.7–10 Radiographically, hamartomas are typically found incidentally, usually in patients between 40 and 60 years of age; nevertheless, pediatric cases have also been reported.10 They are only rarely multiple. Hamartomas may coexist with primary or secondary malignancies of the lung.11,12 Under these circumstances, the hamartomatous lesions may be mistaken for intrapulmonary metastases or synchronous primary carcinomas, particularly if they lack internal calcification.
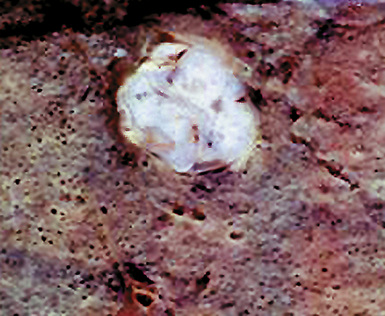
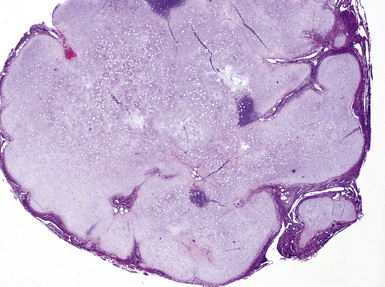
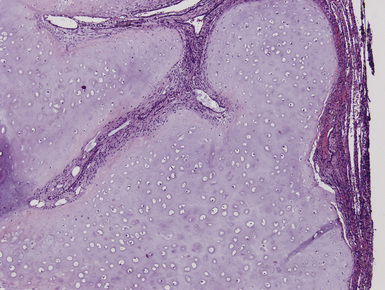
Macroscopically, most PHs are peripherally located, and they sometimes show a topographical relationship to small bronchi or bronchioles.13–16 They rarely penetrate the visceral pleura.17 They are usually well-demarcated (Figs. 18-1 and 18-2) and range from several millimeters to 20 cm in diameter. The central form of hamartoma is encountered in association with large bronchi, as an endoluminal polypoid protuberance covered by intact mucosa.10 All hamartomas of the lung are lobulated, and their cut surfaces reflect their constituent cells (Figs. 18-3 and 18-4). Most of these lesions contain predominantly cartilaginous tissue and are therefore firm to hard, relatively homogeneous, and translucent when transected.

On histologic examination, PHs are manifested by mature mesenchymal tissues but with abnormal configurations. These elements are usually represented by hyaline cartilage, but fibrous tissue, smooth muscle, adipocytic components, and bone18 may be seen. Those masses lacking chondroid elements have sometimes been diagnosed as “intrabronchial lipoma,” “myxoma,” “leiomyoma,” or “fibroadenoma.”13–16 During the growth of PHs, the mesenchymal portions of the lesion engulf and trap small tubular airways, and the latter structures thus spuriously appear to represent an integral part of the lesion (Figs. 18-5 and 18-6). Entrapped alveolar epithelium may also undergo cuboidal or low-columnar metaplasia. Epithelial hyperplasia and papillae may be present in the entrapped epithelium (Fig. 18-7). Indeed, the last of these changes can be prominent and bear a resemblance to placental tissue,19 a feature referred to as “placental transmogrification.” The presence of entrapped epithelium is one of the key distinguishing features of PH from “true” pulmonary chondroma. Transthoracic fine needle aspiration biopsy (FNAB) is a common method for the initial pathologic sampling of mass lesions in the lung. In fact, if PH is the favored diagnosis of the radiologist, FNAB is usually done with the anticipation that a thoracotomy can be avoided if that interpretation is correct. The cytologic features of PH include the presence of dispersed fusiform and stellate cells in a myxoid background, as seen histologically (see Fig. 18-7B). Chondroid material is also present in a majority of cases.20–22
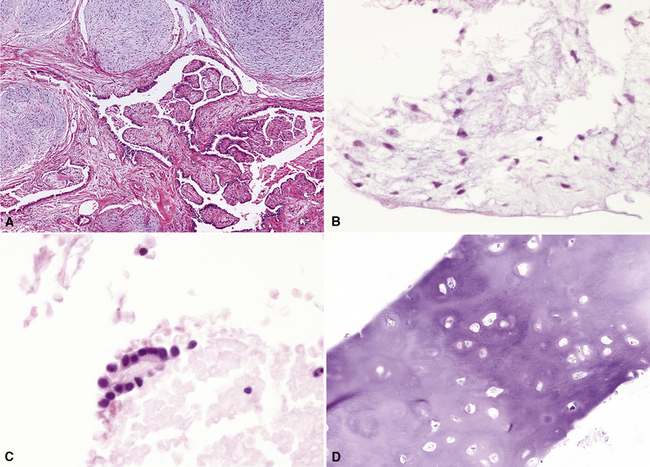
Nevertheless, FNAB of PH may, in fact, result in the very outcome it intends to avoid: namely, formal surgical resection because of a suspicion of malignancy. Some cytologic specimens of PH show atypical epithelial cells with enlarged nuclei, mimicking low-grade adenocarcinoma (see Fig. 18-7C), sometimes adjacent to stromal tissues resembling the pattern seen in some fibroadenomas of the breast in FNAB samples. This can easily lead to a false-positive interpretation of carcinoma.23 Surfactant-containing intranuclear inclusions can be seen in the trapped epithelium of PH,24 like those observed in bronchioloalveolar carcinomas, further suggesting the diagnosis of carcinoma.
Electron microscopic studies of PH show primitive stellate fibroblastic cells with transitional forms to cartilaginous foci.25,26
Cytogenetic evaluations have demonstrated an abnormal karyotype in several instances, principally represented by an exchange of material among various chromosomes.27–30 These data beg the question of whether PH might actually be neoplastic after all, but the clinical evolution of this lesion (see subsequent discussion) generally speaks against such a possibility.
PH must be distinguished from primary or metastatic mesenchymal malignancies in the lungs, as well as primary or secondary biphasic sarcomatoid carcinomas.31–34 The presence of multinucleated cells, nuclear pleomorphism, necrosis, and mitotic activity is rare in PH, in contrast to these diagnostic alternatives. In histologically similar biphasic carcinomas (Fig. 18-8), keratin immunoreactivity is present both in the mesenchymal-like and the overtly epithelial components, whereas PH shows reactivity for epithelial markers only in trapped epithelial elements. True chondromas of the lung tend to be multiple and comprised of only cartilage, which is smoothly circumscribed. In contrast, PHs occur singly, and contain multiple tissue types and entrapped, invaginated epithelium.
PHs continue to enlarge slowly if left in place after diagnosis, but they rarely cause significant clinical difficulty. Simple excision of the lesions is typically performed today, especially with the availability of thoracoscopic video-assisted surgical techniques.35 Laser treatment has also been offered for patients with central PHs.36 The clinical result of these treatments is excellent in virtually all cases.
Returning to the question of whether PH might be neoplastic, Pelosi and colleagues have reported exceptional cases in which a malignancy appears to evolve from this lesion.37 In particular, these authors observed the patterns of malignant mixed tumor and malignant myoepithelioma in association with PH.
Inflammatory Pseudotumor—Plasma Cell Granuloma of the Lung
The most common form of the proliferative spindle cell lesion known as inflammatory pseudotumor (IPT) of the lung has undergone scrutiny in the recent past and is generally regarded currently as a true neoplasm composed of myofibroblasts.2,3 That particular entity had been called the fibrohistiocytic subtype of pulmonary pseudotumor38–40 but has now been renamed inflammatory myofibroblastic tumor (IMT).3 The lesion known as calcifying fibrous pseudotumor41–44 may represent a closely related entity, at least in some cases, and is bound to IMT by its common manifestation of a t(2;17) chromosomal translocation and a potential expression of the ALK-1 protein.45–47 Other lesions of the lung that have been included in the category of IPT—namely, plasma cell granuloma and hyalinizing granuloma46–54—probably do represent non-neoplastic masses with variable etiologies. Both of them are composed of inflammatory and mesenchymal cells, potentially including mature lymphocytes, plasma cells, mast cells, macrophages, eosinophils, fibroblasts, and myofibroblasts. There is also likely overlap between some of these lesions and those described in association with IgG4 sclerosing disease (see later discussion).
As just defined, the true incidence of pulmonary IPTs that are not IMTs is uncertain. IPTs are not commonly encountered in general surgical pathology practice, but their frequency is somewhat dependent on definitions. Some observers have used IPT broadly, to describe both circumscribed nodules and large irregular inflammatory masses, or segmental and lobar consolidations,55 whereas others have almost abandoned the term altogether, preferring a more descriptive diagnosis for most cases, such as organizing pneumonia.
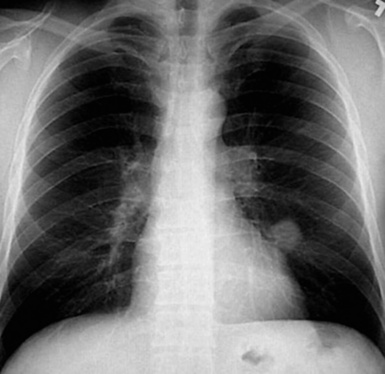
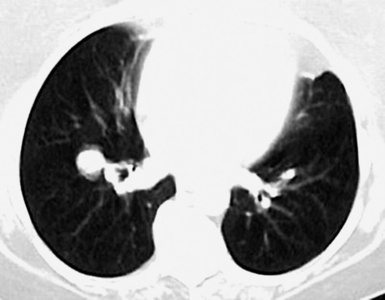
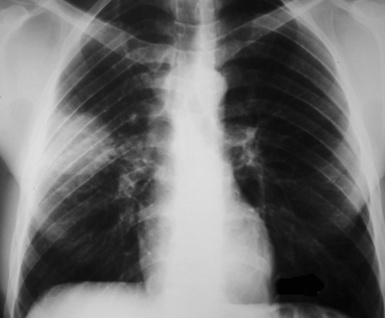
This lesion shows no sex predilection and occurs over a broad age range, from 1 to 77 years, with a mean of 27 to 50 years.40 Approximately 50% of patients complain of cough, hemoptysis, shortness of breath, chest pain, or combinations thereof. Chest radiographs usually show a single, sharply marginated, round or oval mass (Fig. 18-9), but the edges of large lesions may be more ill-defined.56,57 Some IPTs involve the pleural surface and retract it as seen on computed tomography (CT) of the thorax57; as expected, these findings may falsely suggest the possibility of malignancy. Calcification and cavitation also are potentially present in IPTs, and these features may also be present in imaging.
Pulmonary IPTs range in size from 0.5 to larger than 30 cm.39,40 Most have well-defined margins macroscopically but do not have a true fibrous capsule, and the color and texture is variable. Those that contain numerous inflammatory cells are tan-white and fleshy, and those with a predominance of mesenchymal tissue are gray and firm. IPTs with secondary xanthomatization may be bright yellow and friable. Some IPT also exhibit areas of hemorrhage, necrosis, and/or calcification. Rarely IPTs are comprised of sessile intrabronchial masses, whereas others are attached to the pleura. In typical pulmonary IPT, the microscopic architecture of the lung is replaced by a fibroinflammatory proliferation. Depending on their dominant cellular elements and major growth patterns, IPTs may be subclassified into two types: namely, tumefactive organizing pneumonia-like and lymphoplasmacytic variants.39 These may simply represent different stages in the evolution of IPT, but recent publications suggest that lymphoplasmacytic IPT (LPIPT) is distinctive as part of systemic fibrosing autoimmune disorders that feature the presence of numerous IgG4-producing plasma cells.58–61
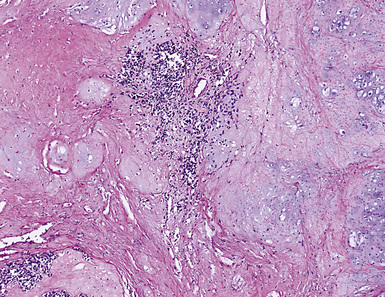
The organizing pneumonia-like variant shows intra-alveolar lymphohistiocytic inflammation and peripheral as well as central fibrosis (Figs. 18-10 and 18-11). Fibroblastic proliferation is admixed with fibrinoinflammatory exudate in alveoli, alveolar ducts, and bronchioles. The alveolar architecture is preserved in early lesions and the peripheral portions of “mature” IPT but is generally obscured by superimposed fibrous tissue, which tends to assume a whorled configuration (Figs. 18-12 and 18-13). Neutrophils are sometimes interspersed with the lymphocytes and plasma cells, and they may form intralesional microabscesses that result in small areas of cavitation. Alveoli bordering IPTs are often filled with foamy macrophages and mantled by hyperplastic pneumocytes. Multinucleated cells of the Touton type are sometimes present, as are foci of dystrophic calcification, osseous metaplasia, or myxomatous change. Lipoid pneumonia may develop adjacent to areas where IPTs have caused bronchial obstruction by intraluminal proliferation or impingement on an airway. Late in the evolution of IPTs and in their central aspects, the lung parenchyma is replaced by deposition of mature collagen in broad bundles that transect the lesion; it may sometimes assume a keloidal appearance.
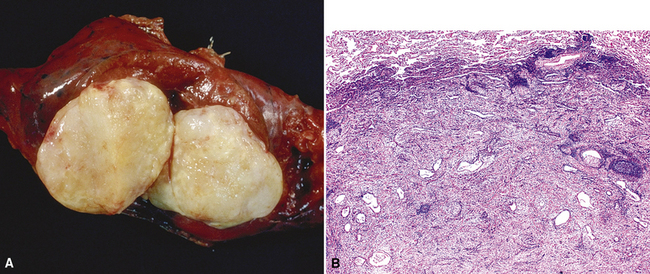
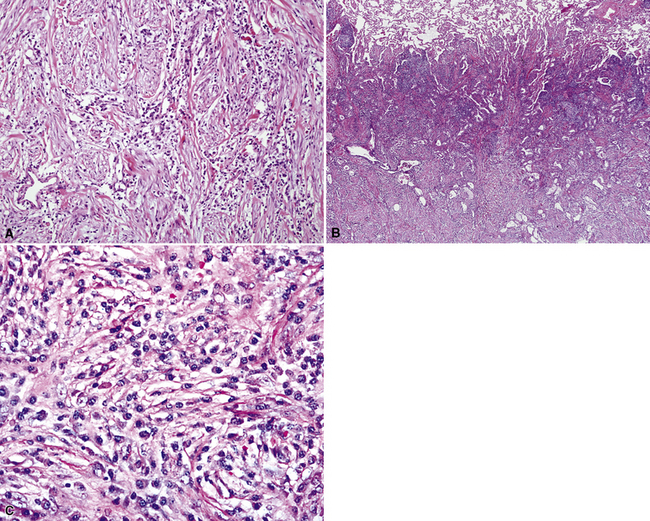
Figure 18-11 A to C, Marked chronic inflammation—with numerous plasma cells—and sclerosis in pulmonary inflammatory pseudotumor.
In the LPIPT variant, plasma cells and lymphocytes comprise the bulk of the lesion; germinal centers and a paucicellular collagenous matrix may also be prominent.58–61 Fibroblasts and xanthoma cells are usually relatively scant. To some degree, the two histologic subtypes of IPT do overlap one another morphologically. As mentioned earlier, the immunohistologic presence of numerous IgG4-positive plasmacytes tends to strongly favor a diagnosis of LPIPT. Other findings that may suggest LPIPT are the presence of endothelialitis, prominent organization, lymphangitic inflammatory infiltrates that are rich in plasma cells and histiocytes (with or without the presence of a mass), and fibrinous pleuritis. Prominently dilated lymphatic spaces, containing histiocytes that show emperipolesis of lymphocytes, may also be observed.
Neither form of IPT is composed of solid sheets of lymphocytes or plasma cells, tending to prevent diagnostic confusion with lymphoma or plasmacytoma. Nevertheless, rearrangement of the immunoglobulin heavy chain genes in a subset of LPIPT cases has been reported, raising the possibility that these particular lesions might be neoplastic or preneoplastic.62
Lymphocytic infiltration and scarring of vascular walls in some examples of IPT, often in association with organizing thrombi, have also been described.39 These changes may be secondary rather than a reflection of a primary vasculitic process. Usually, both kappa and lambda light chain immunoglobulins are detectable immunohistologically in the plasma cells, indicating a polytypic population51; lymphocyte subset markers similarly show an admixture of B cells and T cells.3
The specific etiologic factors underlying the development of pulmonary IPT are largely unknown. The premise that some cases represent a peculiar form of localized pneumonia has support from a history of a previous febrile illness with respiratory complaints in up to 40% of cases. Some case reports have suggested that there is an overlap in appearance between IPT and tumefactive pulmonary infections with aspergillus, rickettsiae, mycoplasma, various viruses, mycobacteria, Cryptococcus, corynebacteria, and other microorganisms.55–57,63–69 Rare examples have also been documented after trauma to the lung,70 and some cases arise from prior aspiration. As stated earlier, current hypotheses hold that some LPIPT are probably part of a systemic autoimmune process.58–61
The differential diagnosis of pulmonary IPT has been partially cited previously. It includes plasmacytoma,71 malignant lymphoma,72,73 and lymphoid hyperplasia,74 selected examples of sclerosing hemangioma of the lung (called epithelial plasma cell granuloma-like tumors by Michal and Mukensnabl75), the peculiar variant of lung cancer known as inflammatory sarcomatoid carcinoma76 (see Chapter 14), and IMT.2,3 Among those conditions, plasmacytomas are recognized by their monotypism for cytoplasmic immunoglobulin, and selected lymphomas may also demonstrate this characteristic. Furthermore, malignant lymphomas are generally less well-circumscribed than IPTs and exhibit more cytologically monotonous infiltrates of atypical lymphoid cells. Localized and diffuse forms of pulmonary lymphoid hyperplasia are composed predominantly of mature lymphocytes, in contrast to the heterogeneous cellular composition of IPTs. Inflammatory sarcomatoid carcinoma can be separated from inflammatory simulators by its diffuse immunoreactivity for keratin, and IMTs contain significantly fewer IgG4-positive cells than those seen in IPTs.60 There is some minor difference of opinion as to whether pulmonary hyalinizing granuloma77 is a part of the spectrum of IPT in the lung. Pulmonary hyalinizing granuloma shows more lamellar hyalinized collagen than is seen in classical IPT. Hyalinizing granulomas of the lung are commonly multiple, whereas “usual” IPT is not.77 Sclerosing hemangiomas78 were once considered to be related to IPTs, but they are now appreciated as epithelial neoplasms with pneumocytic differentiation.79 The latter lesions may exhibit sclerosis, but they also contain aggregates of bland cuboidal cells together with micropapillary and angiomatoid areas. Inflammation is absent or scanty in sclerosing hemangiomas, and their constituent cells express thyroid transcription factor-179; those of IPT do not. Ledet and associates80 have examined the utility of immunostains for mutant p53 protein in the diagnostic separation of IPTs from low-grade intrapulmonary sarcomas. In their hands, p53 was restricted to malignant lesions, albeit with less than absolute sensitivity for such tumors.
Some examples of pulmonary IPT have been monitored for extended periods of time before excision or autopsy examination.81–83 Information from these cases indicates that the lesions tend to remain stable or grow very slowly. Spontaneous resolution has also been documented, and a few lesions have shrunk after small incisional biopsy or administration of systemic corticosteroids or irradiation.81,83 Surgical removal is usually necessary to establish a definitive diagnosis of IPT, and, if the lesion has been completely excised, no further therapy is needed.56 Long-term follow-up of patients with pulmonary IPTs has revealed no untoward clinical events in such cases.
Mycobacterial Spindle Cell Pseudotumor
Spindle cell pseudotumors that are reactions to mycobacterial infection have been documented in several organ sites in immunosuppressed patients.84–86 These proliferations show a close histologic resemblance to histoid leprosy,87,88 and most reports have documented numerous intralesional mycobacteria (see Chapter 6). Only one case of mycobacterial pseudotumor (MP) has been reported in the lung,89 although we have anecdotally encountered another example in a 41-year-old male patient with acquired immunodeficiency syndrome (AIDS).
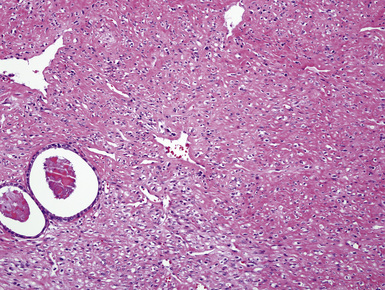
Grossly, the lesions appear as yellow-gray nodules. They may show a predilection for small airways. Microscopically, the lesions are comprised of aggregates of spindle cells with a fascicular growth pattern and without significant atypia or mitoses. Scattered lymphocytes and plasma cells may be present, but overt granulomas are lacking. The cytoplasm of the spindle cells is “foamy” but may contain hemosiderin focally. The lesional cells are immunoreactive for lysozyme, with no labeling for S100 protein, keratin, actin, desmin, or von Willebrand factor. Ziehl-Neelsen staining shows innumerable acid-fast bacilli in the fusiform cells (Figs. 18-14 to 18-16). Most examples of MP in other anatomic locations have been related to Mycobacterium avium-intracellulare or Mycobacterium kansasii.
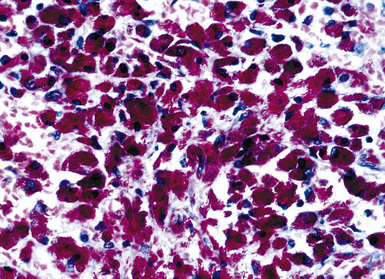
Another reported feature of MPs in other sites is a possible source of diagnostic error. That is, a reproducible cross-reaction has been seen with mycobacterial antigens using certain desmin antibodies,90 spuriously suggesting the presence of a myogenous proliferation. This observation is especially troublesome in the setting being discussed here, because smooth muscle or myofibroblastic tumors enter prominently into the differential diagnosis of MPs. However, a documented lack of immunoreactivity for actin and electron microscopic attributes that support histiocytic differentiation in MP argue against those alternative interpretations.
Other lesions that must be separated from pulmonary MP include Kaposi sarcoma, malignant fibrous histiocytoma, spindle cell melanoma, and neural proliferations.89 Obviously, acid-fast stains should be done in all spindle cell lesions from immunocompromised individuals, and these consistently confirm mycobacterial causation. In some instances, the nature of MP is more obvious because of overtly granulomatous foci in the lung tissue around the spindle cell lesion. Characteristics of malignancy such as necrosis, nuclear atypia, and pathologic mitoses are absent in MP.84–86 Thus, diagnoses of pulmonary sarcomatoid carcinoma, malignant fibrous histiocytoma, or other sarcomas would be unlikely.
Pseudoneoplastic Hematolymphoid Processes
Lymphoid interstitial pneumonia and nodular lymphoid hyperplasia may both be regarded as pseudoneoplasms. They are discussed in Chapter 15.
Rosai-Dorfman Disease (Sinus Histiocytosis with Massive Lymphadenopathy)
Most frequently, Rosai-Dorfman disease (sinus histiocytosis with massive lymphadenopathy [SHML]) involves lymph nodes, but it may rarely involve the lung primarily.91,92 It can be seen in either sex and over a wide range of ages. Despite the name of this condition, lymphadenopathy does not always coexist with extranodal disease. When this condition affects the pulmonary parenchyma, it appears to have its epicenter in the hilar tissue and follows lymphatics peripherally into both lungs.91,93 Accordingly, chest radiographs show an accentuation of central bronchovascular markings and bilateral interstitial prominence. CT scans (Fig. 18-17) may show areas of pleural-based consolidation. Confluence of the infiltrates may yield masslike densities in the lung fields as well (Fig. 18-18). Interestingly, clinical evidence of associated immune dysfunction may be apparent, including autoantibody formation, polyarthritis, immune-complex glomerulonephritis, asthma, and juvenile diabetes mellitus. Fever, night sweats, and weight loss have also been reported.91 Justification for the classification of Rosai-Dorfman disease as non-neoplastic comes from molecular data indicating its polyclonal nature.91 Nevertheless, this condition may occasionally coexist with solid malignancies, including carcinomas of the lung.94,95 Such an association could arise, at least in part, from the aforementioned immunologic dysfunction in SHML.
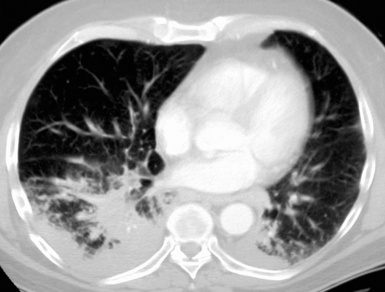
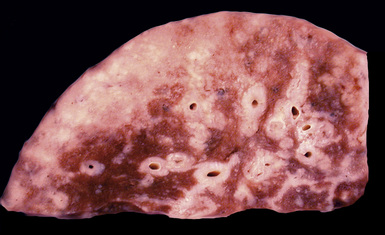
Figure 18-18 Gross photograph of lung tissue in Rosai-Dorfman disease (from the same patient as Fig. 18-17). Note the lymphangitic pattern of involvement.
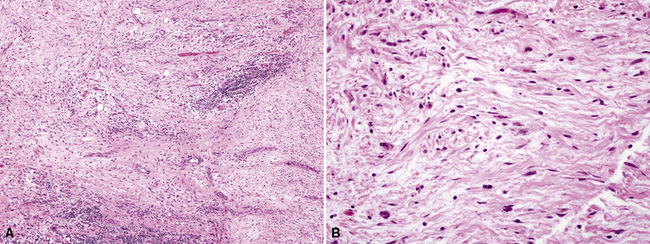
Pathologic specimens of the lung or lymph nodes in SHML demonstrate comparable morphologic findings. Dilated lymphatic spaces in the pulmonary parenchyma contain large pale histiocytes with abundant amphophilic cytoplasm, surrounded by lymphoid infiltrates that are punctuated by germinal centers and fibrous septa (Figs. 18-19 and 18-20). The large histiocytes demonstrate a peculiar tendency to engulf intact, mature lymphocytes, representing a phenomenon known as lymphemperipolesis (Fig. 18-21
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


