Figure 34–1.
Cardiac phenotype of PFHBI patients. (A) ECG from the large Afrikaner family. (a) Sinus rhythm with a RBBB in an 8-year-old asymptomatic boy on a standard 12-lead ECG, with leads Std I, V1, and V6 shown. (b) ECG showing 2:1 atrioventricular node block (atrial rate, 76 bpm; ventricular rate, 38 bpm) with a broad QRS complex on Holter monitoring in a 54-year-old man who had recently become symptomatic. (B) ECG of the Lebanese family showing a complete RBBB (cRBBB) with a right-axis deviation (From Kruse et al. [93]. With permission from American Society for Clinical Investigation)
In 1978, Stephan [27] reported a large Lebanese family. Among the 209 patients included in the study, 31 were diagnosed with conduction defects and three were implanted with a pacemaker. Within the family, conduction defects were mostly RBBB (12 patients), incomplete RBBB (seven patients), RBBB with left axis deviation (six patients), RBBB with right axis deviation (four patients) and complete AV block (two patients). The same authors reported in 1997 a follow-up of a family spanning five generations composed of 47 patients with major CDD and 36 patients with minor CCD (Fig. 34.1b) [28]. Conduction defects in this family were diagnosed early in life and were progressive in 5–15 % of the patients evolving to complete AV block.
In 1995, the disease locus in both the Lebanese and the South African family reported previously was mapped to chromosome 19q13.2-13.3 [29, 30]. The penetrance of the disease appears variable in the Lebanese families whereas almost complete in the South African family.
In 2009, Kruse et al. [93] identified the disease causing mutation in the large South African Family in TRPM4 gene. The mutation (TRPM4 c.19G->A) predicted the amino acid substitution p.E7K in the TRPM4 amino terminus. This gene encodes a member of the melastatin subfamily of transient receptor potential (TRP) channels. The encoded protein is a monovalent-permeable, nonselective cation (CAN) channel [31, 32] and comprises six predicted transmembrane spanning helices (TM1–TM6), a cytoplasmic N- and C-terminal domain, and a pore (P) region between TM5 and TM6, thus, resembling the membrane topology of voltage-gated cation channels [33]. Although this channel is regulated by (Ca2+), it is not permeable for Ca2+. The opening probability of the TRPM4 channel increases during depolarization because of an intrinsic voltage-sensing mechanism [34]. TRPM4 channels are a likely molecular correlate of the cardiacCa2+-activated CAN channel, which plays a key role in regulating the driving force for Ca2+ entry in cardiac cells [31, 35]. Quantitative analysis of TRPM4 mRNA content in human cardiac tissue showed the highest expression level in Purkinje fibers. Cellular expression studies showed that the c.19G->A missense mutation attenuated deSUMOylation of the TRPM4 channel. The resulting constitutive SUMOylation of the mutant TRPM4 channel impaired endocytosis and led to elevated TRPM4 channel density at the cell surface corresponding to a gain-of-function mechanism underlying this type of familial heart block.
In 2010 Lui et al. identified the causal gene in the large Lebanese family in the same TRMP4 gene (p.Ala432Thr) and reported two additional mutations in two smaller French families (p.Arg164Trp, and p.Gly844Asp) [36]. Altogether, clinical features in the three families were: two cases of LAD, ten cases of complete RBBB, 16 cases of incomplete RBBB, 15 cases of complete RBBB with LAD, six cases of complete RBBB with RAD, seven cases of AVBc and no cases of isolated RAD. A remarkable feature in these three families was that no family members had an incomplete or complete left bundle-branch block (LBBB). All three mutations result in an increased current density. This gain of function is due to an elevated TRPM4 channel density at the cell surface secondary to impaired endocytosis and deregulation of SUMOylation.
All together these findings highlight endocytosis as a new mechanism of ion channel density control for electrical disturbances in the heart.
Following the identification of mutations in the TRPM4 gene in the four pedigrees described above, Stallmeyer et al. screened 160 unrelated patients with various types of inherited cardiac arrhythmic syndromes [37]. Among the 45 cases with RBBB or AVB eight rare TRPM4 variant were indentified. RBBB appeared to be the leading electrocardiographic phenotype (26.3 %) and 11.5 % of cases with AVB were associated with rare TRMP4 variant. Among the eight variant, two (p.G844D and A.432 T) have been previously identified by Hui et al.; five segregated in small families and three were identified in sporadic cases. In two families the probands presented congenital conduction defects (p.K914R; p.Y790H) whereas 3 families presented a progressive form of conduction defect (p.P970S; p.G844D). In all cases TRMP4 mutations were associated with a wide spectrum of severity. Of interest p.G844D mutations has now been reported in one French and two German unrelated families and no TRPM4 mutations were identified in patients presenting sinus node dysfunction, Brugada syndrome, or long-QT syndrome.
Cardiac Specific Sodium Channel Nav1.5 Causes Progressive Cardiac Conduction Defect
In 1999, Schott et al. reported an independent large multigenerational pedigree presenting PCCD [38] and identified a mutation in the cardiac sodium channel Nav1.5.
Familial and clinical investigation in the large Lenègre pedigree started after the identification of a member with right bundle branch block (RBBB) and syncope, a brother with RBBB and a sister with complete AV block and syncope. Among the 65 family members included in the study, 15 had clinical and electrocardiographic abnormalities [39]. Familial investigation found all types of CCD in the family (Fig. 34.2). RBBB was present in five, left bundle branch block (LBBB) in two, left anterior or posterior hemiblock in three, and long PR interval (more than 210 ms) in eight members. None had a structural heart disease. Five members had received a pacemaker implantation because of syncope or complete AV block. A typical example of progressive development of conduction defect in a same patient is shown in Fig. 34.3. Electrocardiograms recorded in 1982, 1998, and 2000 show progressive increase in QRS duration (QRS: 130, 140, and 172 ms).
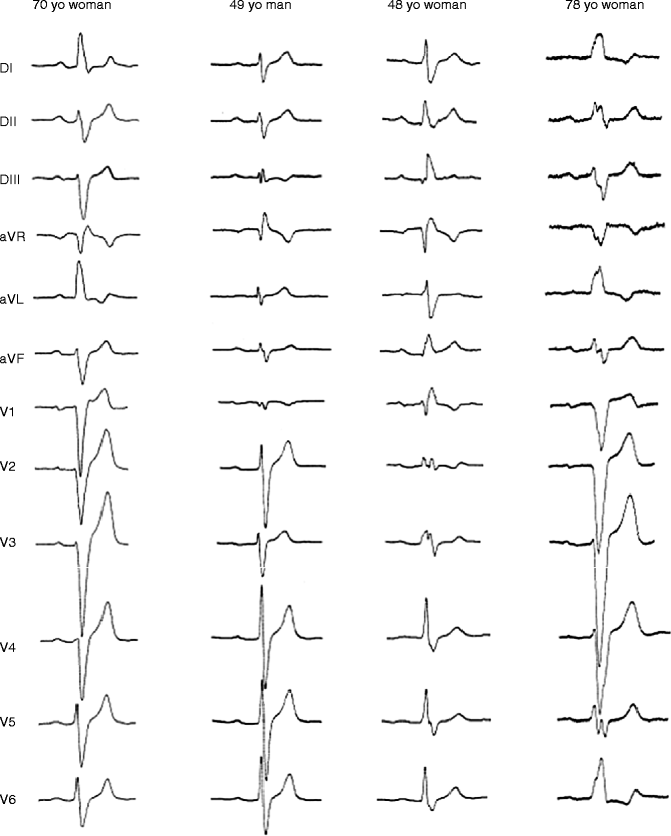
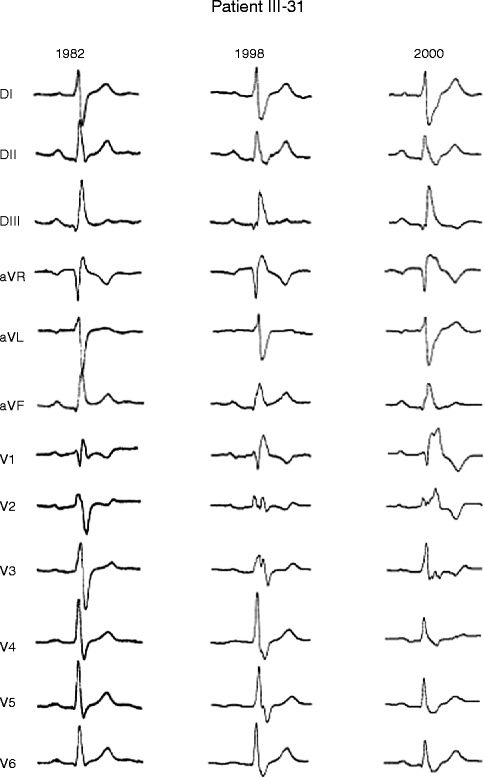
Figure 34–2.
Examples of electrocardiogram patterns in SCN5A affected members. Patient I was a 70-year-old woman with parietal block and left-axis deviation (heart rate [HR]: 64 beats/min, P: 144 ms, PR: 215 ms, QRS: 160 ms). A pacemaker was implanted because of several episodes of syncope. Patient 2 was a 49-year-old man with a parietal block and undetermined axis (HR: 54 beats/min, P: 150 ms, PR: 244 ms, QRS 128 ms). Patient 3 was a 48-year-old woman with right bundle branch block (HR: 64 beats/min, P: 153 ms, PR: 205 ms, QRS: 172 ms). Sudden widening of QRS complexes and occurrence of 2–1 AV block during exercise led to pacemaker implantation. Patient 4 was a 78-year-old woman with left bundle branch block (HR: 59 beats/min, P: 157 ms, PR: 248 ms, QRS: 196 ms). A pacemaker was implanted after two episodes of syncope
Figure 34–3.
Serial electrocardiograms performed in 48-year-old woman SCN5A positive PCCD patient showing progressive development of conduction defect. Electrocardiograms recorded in 1982, 1998, and 2000 show progressive increase in QRS duration (QRS: 130, 140, and 172 ms)
Long-term follow-up of several affected members demonstrated that their conduction defects increased in severity with age. The age of the patients who participated to the study ranged from 15 to 81 years. We plotted conduction parameters in relation to age (Fig. 34.4a). Whatever the age, averaged and filtered P-wave, PR, and QRS duration were longer in affected patients than in unaffected. There was a shift in the regression line for P-wave and PR duration toward higher values in affected patients, whereas the slopes, expressed in ms per year, were comparable in the two groups. In contrast, the QRS duration evolved differently in relation to age between the two groups. There was a more pronounced QRS lengthening with age in affected than in unaffected patients. In addition, an age-dependent variability in the QRS duration was evidenced in the affected group.
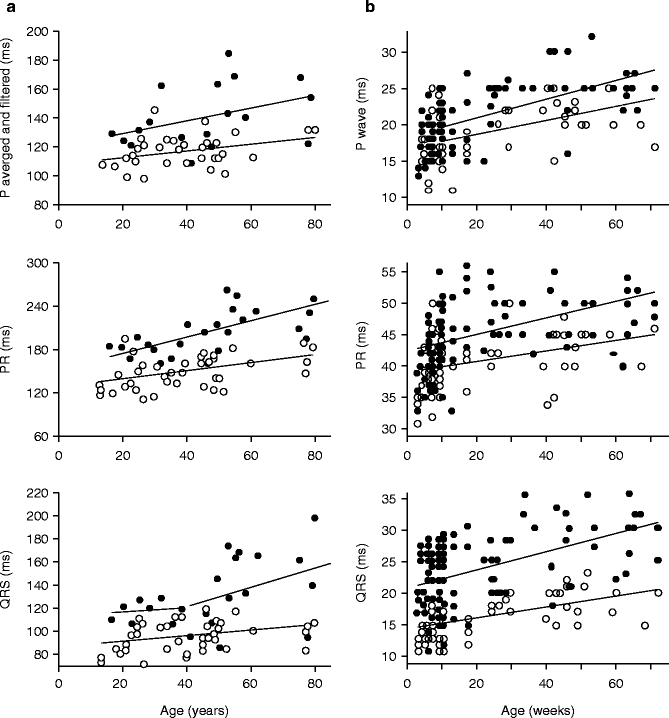
Figure 34–4.
Evolution with aging of conduction parameters in humans and in a scn5a+/− knock-out mouse model. (a) Evolution with aging of conduction parameters in affected (filled symbols) and unaffected subjects (open symbols). Top panel: Averaged and filtered P-wave duration. Middle panel: PR duration. Bottom panel: QRS duration. In top and middle panel, data were fitted with a linear regression analysis. In bottom panel, assessment of the residuals showed that the linear model was poorly adapted to fit the relation between QRS duration and aging in affected members. The variance was significantly different before and after the age of 40 (ratio variance test; p < 0.001) was indicative of a threshold effect of age. (b) Effects of age on P-wave duration, PR interval duration, and QRS interval duration in WT (open symbols) and Scn5a+/− (filled symbols) mice
These data demonstrate that conduction was already abnormal early in life in the absence of specific conduction defects, which are never observed before the age 40 years. In the family, young affected patients have ECG parameters considered within ‘normal limits’. Using the presence of selective conduction defects as a selective criteria, penetrance is compete only late during aging.
Molecular genetics excluded the chromosome 19 locus and linkage analysis mapped the disease locus on chromosome 3 near SCN5A gene encoding the cardiac specific sodium channel Nav1.5. SCN5A was considered as a candidate gene and direct sequencing of affected members identified a splice donor site mutation in exon 22 of SCN5A gene (IVS.22+2T->C) in 25 affected members [38]. The abnormal transcript is predicted to an in-frame skipping of exon 22 and an impaired gene product missing the voltage-sensitive DIIIS4 segment. In vitro exon-trapping experiments of the mutated SCN5A IVS.22+2T->C gene confirmed skipping of exon 22 [41]. Wild-type and exon 22-deleted Nav1.5 channels were expressed in COS-7 cells. Current traces show that Lenègre disease results from a loss-of-function mutation disease. Immunostaining in cells transfected with exon 22-deleted SCN5A suggests that the protein is correctly processed to the cell membrane.
Together with the first description of Lenègre-disease SCN5A-causing mutation, a second SCN5A-5280 del G frameshift mutation was described in a Dutch family with non-progressive conduction defect [39]. The proband presented after birth with an asymptomatic first-degree AV block associated with RBBB (PR interval and QRS duration: 200 and 120 ms, respectively). Three brothers were asymptomatic, one of which with RBBB (QRS duration: 110 ms). The asymptomatic mother had an unspecified conduction defect (QRS duration: 120 ms).
Altogether, a supposed 50 % reduction of sodium current is tolerated to some extent. The effect of the mutation only becomes evident later in age, when conduction in the heart becomes impaired because of the naturally occurring aging process. Interestingly the normal aging process usually involves sclerosis, although evidence is emerging (as described in the next section of this chapter) that sclerosis is enhanced in carriers of loss-of function SCN5A mutations.
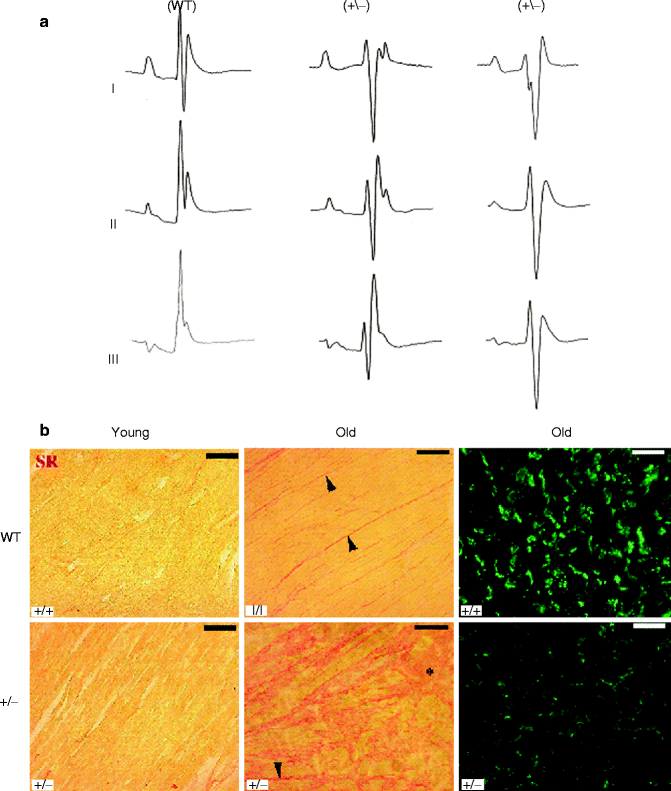
Figure 34–5.
Surface ECG recordings and histology on WT and Scn5A +/− mice. (a) Surface ECGs in WT and Scn5a+/− mice. Representative ECG recordings (leads I, II, and III) in a WT and two different Scn5a+/− mice. (b) Ventricular fibrosis and expression of connexin 43. Ventricular fibrosis (in red) in young mice (left) is virtually absent in WT, which leaves the expression pattern of Cx43 unaffected. Fibrosis in old WT animals is increased in the interstitium between the muscle fibers whereas locally massive fibrosis is observed in Scn5A+/− old animals. Fibrosis is accompanied by a down regulation and redistribution of Cx43 in old Scn5A+/− mice, whereas Cx43 in old WT hearts appears unaffected. Bar = 50 μm in pictures showing Cx43 and 100 μm in those showing Sirius red
A Scn5a+/− Mouse Model Mimics Lenègre Disease
A mouse model with targeted disruption of Scn5a has been established by Andrew Grace’s group from the University of Cambridge [40]. In this model, exon 2 of SCN5A gene was replaced with a splice acceptor (SA)-Gfp-PGK-neomycin cassette. Homozygous knockout mice die during embryonic development due to severe defects in ventricular morphogenesis whereas heterozygote (Scn5a+/−) mice show normal survival. In the initial report, it was shown that INa was reduced by about 50 % in isolated ventricular myocytes from 8 to 10 week-old heterozygote mice as compared to wild-type mice. It was also shown that 8–10 week-old Scn5a+/− mice have several cardiac defects including decreased atrial and atrio-ventricular conduction, as well as increased inducibility of ventricular arrhythmias. Following this initial report, the model was further analyzed to investigate the potential pathophysiological mechanisms involved in SCN5A-related PCCD [41, 42, 43]. ECG recording and activation mapping on Langendorff-perfused hearts in 4–71 weeks old mice showed that ventricular conduction was also decreased and that the ventricular phenotype worsened with age (Fig. 34.4b). In young Scn5a+/− mice, conduction velocity was only affected in the right ventricle. In old mice, right ventricular conduction defect worsened and was in addition associated with conduction defect in the left ventricle. The age-dependent increase in conduction defect was associated with the occurrence of fibrosis in ventricular myocardium. In some aspects, this phenotype resembles Lenègre-Lev disease, although in the later fibrosis was found to be limited to the conduction system area [44]. Whether fibrosis is a direct consequence of Nav1.5 loss of function or results from desynchronized ventricular contractile activity is still an open question. Whatever its triggering mechanism is, fibrosis, as well as associated redistribution of connexin 43 expression, most likely contribute to the age-dependent degradation of conduction in the mouse model (Fig. 34.5). The hypothesis that SCN5A-related inherited PCCD in human also results from a primary decrease in Na+ current and a secondary progressive fibrosis with aging is unlikely to be clarified, but it is worth noting that fibrosis has been found in patients with the SCN5A-related Brugada syndrome [45, 46]. In this context, Scn5a+/− mice represent a promising tool for testing preventive therapies as an alternative to pacemaker implantation.
Other Familial Forms of Cardiac Conduction Defects Linked to SCN5A Mutations
Since its first description in 1999, many new SCN5A mutations causing progressive or congenital CCD have identified, sometimes associated with dilated cardiomyopathy or arrhythmias.
In 2001, Tan et al. [47] provided a functional characterization of a SCN5A mutation that causes a sustained, isolated conduction defect with pathological slowing of the cardiac rhythm. ECG findings reported bradycardia, AV-nodal escape, broad P wave, long PR and wide QRS. By analyzing the SCN5A coding region, they reported a SCN5A-G514C mutation in five affected family members. Biophysical characterization of the mutant channel showed abnormal voltage-dependent ‘gating’ behavior.
In 2002, Wang et al. [48] reported two new SCN5A mutations that result in AV conduction block presented during childhood. Molecular genetic studies revealed a first SCN5A-G298S mutation in a proband with progressive AV block (QRS = 135 ms at age 9 and QRS = 133 ms at age 20). A second SCN5A-D1595N mutation was identified in a proband with complete heart block at the age of 12 years. The functional consequences of the two mutations are impaired fast inactivation but not sustained non-inactivating current. The mutations reduce the sodium current density and enhance slow inactivation components.
In 2003, Bezzina et al. [49] reported a family in which the proband born in severe distress with irregular wide complex tachycardia. An older sister died at 1 year of age from severe conduction disease with similarly widened QRS-complexes. Mutational analysis in the proband demonstrated compound heterozygosity for a nonsense SCN5A-W156X mutation, inherited from the father, and a SCN5A-R225W missense mutation, inherited from the mother. Expression studies showed that the W156X mutation is a loss of function mutation whereas the R225W mutation leads to a severe reduction in current density and is also associated with gating changes. Histological examination of the heart from the deceased sibling revealed changes consistent with a dilated type of cardiomyopathy and severe degenerative abnormalities of the specialized conduction system. These morphological changes may have occurred secondary to the sodium channel abnormality and contributed to the severity of the disorder in this individual.
In 2006, Niu et al. [50] reported a novel SCN5A-W1421X mutation in a four-generation family with cardiac conduction abnormalities and several cases of sudden death. Most family members who carry this W1421X mutation have developed major clinical manifestations. However, in a 73-year-old grandfather, who carried the SCN5A-W1421X mutation, a second SCN5A-R1193Q variant was identified. This patient has remained healthy and presents only minor electrocardiographic abnormalities, whereas most of his siblings, who carried a single mutation (W1421X), had died early or had major disease manifestations. This observation suggests that the R1193Q mutation has a complementary role in alleviating the deleterious effects conferred by W1421X in the function of the SCN5A gene. This report provides a good example to explain the mechanism of penetrance of genetic disorders.
The work of Viswanathan et al. [51] provides a second illustration of phenotypic modulation by an SCN5A polymorphism. They identified a novel mutation SCN5A-T512I, in a 2-year-old boy diagnosed with second-degree AV conduction block. The T512I mutation, when heterologously expressed, causes hyperpolarizing shifts in activation and inactivation, and enhances slow inactivation. However, the common SCN5A-H558R polymorphism also found in this child, has no effect on wild-type sodium current but attenuates the gating effects caused by the T512I mutation. The polymorphism entirely restores the voltage-dependent activation and inactivation voltage shifts caused by the T512I mutation, and partially restores the kinetic features of slow inactivation. This mutation and the H558R polymorphism were both found in the same allele of the child with isolated conduction disease, suggesting a direct functional association between a polymorphism and a mutation in the same gene.
Several reports identified SCN5A-D1275N mutation associated with CCD and various cardiac arrhythmias or structural cardiomyopathies. In 2003, Groenewegen et al. [52] reported a SCN5A-D1275N mutation co-segregating with two connexin 40 genotype [(−44 (G–>A) and +71 (A–>G)] in familial atrial standstill (AS). SCN5A-D1275N channels show only a small depolarizing shift in activation compared with wild-type channels. The authors proposed that, although the functional effect of each genetic change is relatively benign, the combined effect of genetic changes eventually progresses to total atrial standstill. More recently, a SCN5A-L212P mutation associated with the same connexin 40 polymorphisms combination was reported in another atrial standstill family [53].
In 2004, McNair et al. [54] reported a large family diagnosed with CCD associated with sinus node dysfunction, arrhythmia, right and occasionally left ventricular dilatation and dysfunction. Screening family members for mutations identified a SCN5A-D1275N defect. This finding expands the clinical spectrum of disorders of the cardiac sodium channel to cardiac dilation and dysfunction and supports the hypothesis that genes encoding ion channels can be implicated in dilated cardiomyopathies. In 2005, Olson et al. [55] reported a SCN5A-D1275N mutation associated to susceptibility to heart failure and atrial fibrillation.
In 2006, Laitinen-Forsblom et al. [56] reported a five family member with atrial arrhythmias and intracardiac conduction defects. Interestingly left ventricle dilatation was also seen in one individual and three individuals had a slightly enlarged right ventricle. Premature death due to stroke occurred in one subject, and two other members had suffered from stroke at a young age. Direct sequencing disclosed a SCN5A-D1275N mutation. This alteration was present not only in all six affected individuals, but also in two young individuals lacking clinical symptoms.
Sodium Channel Beta1 Subunit Mutations Associated with Cardiac Conduction Disease
Regulatory proteins of cardiac sodium channels Nav1.5 are logical candidates for CCD. Sodium channels are multisubunit protein complexes composed not only of pore-forming α subunits but also of multiple other protein partners including auxiliary function-modifying β subunits. The β1 transcript arises from splicing of exons 1–5 of SCN1B gene and a second transcript has been described, arising from splicing of exons 1–3 with retention of a segment of intron 3 (termed exon 3A), leading to an alternate 3’ sequence (β1B transcript).
Watanabe et al. screened 44 probands with conduction disease for mutations in SCN1B and β1B transcript [57]. They reported 1 SCN1B and 2 β1B mutations in three kindred’s with conduction abnormalities.
A missense mutation, c.259G->C in exon 3, resulting in p.Glu87Gln within the extracellular immunoglobulin loop of the protein was identified in a Turkish kindred affected by conduction disease. None of the families had a history of syncope, sudden cardiac death, or epilepsy. The proband was a 50-year-old woman who presented with palpitations and dizziness and had complete left bundle branch block. Electrophysiologic analysis revealed a prolonged His-ventricle interval of 80 ms and inducible atrioventricular nodal reentrant tachycardia; complete atrioventricular block occurred following atrial programmed stimulation and during induced tachycardia. Her brother was found to have bifascicular block (right bundle branch block and left anterior hemiblock), whereas their mother had a normal ECG.
Two nonsense mutations in the same codon were identified in two other families. A c.536G→A in exon 3A in a French kindred affected with Brugada syndrome and conduction disease. This mutation results in p.Trp179X and is predicted to generate a prematurely truncated protein lacking the membrane-spanning segment and intracellular portion of the protein. The French proband was a 53-year-old man who presented with chest pain but had normal coronary angiography and echocardiography. ECG revealed ST segment elevation typical of Brugada syndrome as well as conduction abnormalities, including a prolonged PR interval of 220 ms and left anterior hemiblock. A baseline ECG in his brother, who had no history of palpitations or syncope, showed left anterior hemiblock and minor ST segment elevation suggestive of Brugada syndrome (type II saddleback) abnormalities. Their sister had a normal ECG but her son was found to have right bundle branch block and type II Brugada syndrome after flecainide challenge.
A different nonsense mutation, c.537G→A in exon 3A resulting in p.Trp179X, affecting the same codon as in family 2, was identified in a Dutch kindred. The Dutch proband was a 17-year-old girl who had right bundle branch block and a prolonged PR interval of 196 ms on ECG; echocardiography was normal and a flecainide test for Brugada syndrome was negative. Her father had a normal ECG.
Both the canonical and alternately processed transcripts were expressed in the human heart and were expressed to a greater degree in Purkinje fibers than in heart muscle, consistent with the clinical presentation of conduction disease. Sodium current was lower when Nav1.5 was coexpressed with mutant β1 or β1B subunits than when it was coexpressed with WT subunits. These findings implicate SCN1B as a disease gene for human arrhythmia susceptibility.
Connexin 40 Mutation Associated with a Malignant Variant of Progressive Familial Heart Block Type-1
Cardiac myocyte excitability in atria, His-Purkinje system and ventricles is largely determined by the properties of voltage-gated Na channels. Once activated, excitatory currents rapidly propagate to neighbouring cells via low-resistance intercellular channels called gap junctions, which facilitate the synchronous contraction of the heart. Connexins, or gap junction proteins, are a family of structurally-related transmembrane proteins that assemble to form vertebrate gap junctions. Connexins are four-pass transmembrane proteins with both C and N cytoplasmic termini, a cytoplasmic loop (CL) and two extra-cellular loops, (EL-1) and (EL-2). Connexins are assembled in groups of six to form hemichannels, or connexons, and two hemichannels then combine to form a gap junction.
Makita et al. evaluated 156 probands of diverse ethnic backgrounds with clinical diagnosis of PFHBI and identified a germline heterozygous missense mutation in exon 2 of the Cx40 gene, GJA5.
The proband, an 11-year-old male at time of death, was first referred for evaluation when he was 6 years-old because of ECG abnormalities. Although asymptomatic at that time, his ECG showed advanced atrioventricular (AV) block, complete left bundle branch block (LBBB) and left axis deviation (Fig. 34.6). The proband died suddenly 5 years later during exercise. The proband’s younger sister shares the Cx40-Q58L mutation. She is asymptomatic with a QRS duration at the upper limit of normal, left axis deviation that has been progressive and QRS notch. These findings are consistent with impaired intraventricular conduction. The mother died suddenly at age 30, after delivering the second child. An ECG on record, obtained when she was 16 years old, was similar to that of the proband. In addition, a ventricular tachycardia was recorded during the recovery phase of an exercise stress test.
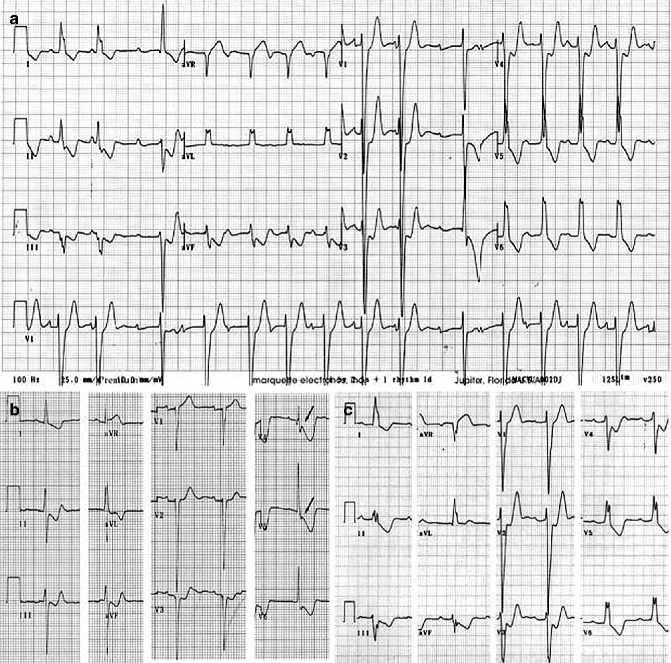
Figure 34–6.
ECG of Connexin 40 PCD patients. (a) ECG of proband at age 6 showing advanced AV block, complete LBBB and left axis deviation. Patient died suddenly 5 years later. (b) ECG of proband’s sister at age 6 showing QRS duration at upper limit of normal, left axis deviation that has been progressive, and QRS notch in V4 and V5 (arrows) consistent with impaired intraventricular conduction. (c) ECG of proband’s mother at age 16 showing complete LBBB and left axis deviation. She died suddenly at age 30
The GJA5 mutation (c.173 A>T) caused an amino acid substitution (glutamine (Q) replaced by leucine (L)) at position58 in Cx40 (Cx40-Q58L) [58]. Heterologous expression experiments revealed that this novel mutation (Cx40-Q58L) significantly impaired the ability of Cx40 to form gap junction channels. Confocal microscopy showed that the Cx40-Q58L mutant but not the wild type (WT) failed to form plaques at sites of cell-cell apposition. Co-expression experiments indicated that Cx40-WT protein provided only partial rescue of the Cx40-Q58L cellular phenotype. Cells expressing both WT/Q58L showed intermediate conductance (15.4 ± 3.7 nS, n = 16) between WT (28.8 ± 3.6 nS, n = 16, p < 0.001) and Q58L (0.28 ± 0.11 nS, n = 14, p < 0.001).
Of interest other studies have shown somatic mutations of Cx40 or Cx43 in patients with idiopathic atrial fibrillation [59, 60] those mutations were confined to the atria, and conduction abnormalities in the ventricles or His-Purkinje system were not observed. A second recent publication by Norstrand et al. associated a de novo missense mutation in connexin 43 (E42K-Cx43) with Sudden Infant Death [61]. The E42K-connexin43 demonstrated a trafficking-independent reduction in junctional coupling, in vitro and a mosaic pattern of mutational DNA distribution in deceased cardiac tissue, suggesting a novel mechanism of connexin 43-associated sudden death.
SCN5A Overlap Syndrome
Cardiac sodium channel dysfunction caused by mutations in the SCN5A gene is associated with a number of rare arrhythmia syndromes, including long-QT syndrome type 3 (LQT3), Brugada syndrome, conduction disease, sinus node dysfunction, and atrial standstill, which potentially lead to fatal arrhythmias in relatively young individuals. Although these various arrhythmia syndromes were originally considered separate entities, recent evidence indicates more overlap in clinical presentation and biophysical defects of associated mutant channels than previously appreciated. Various SCN5A mutations are now known to present with mixed phenotypes, a presentation that has become known as “overlap syndrome of cardiac sodium channelopathy.” In many cases, multiple biophysical defects of single SCN5A mutations are suspected to underlie the overlapping clinical manifestations [62].
One of the first reports on this entity was provided by Bezzina et al. with the description of the SCN5A-1795insD+/− mutation in a large multigenerational family presenting with extensive variability in type and severity of symptoms of sodium channel disease [63]. Heterogeneous biophysical properties of this and other SCN5A mutations underlying the mixed disease expressivity are now increasingly recognized. Other determinants of sodium channel disease expressivity, including genetic background, concomitant disease and environmental factors, are suspected but still largely unknown [64, 65]. Identification of these modifiers will prove essential for improved diagnosis and risk stratification in patients with sodium channelopathies.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


