Fig. 48.1
Mechanisms of proarrhythmia in comparison with normal cardiac action potentials illustrated, with alterations in the cardiac action potential noted by a dotted line. (a) Normal cardiac action potential, with the primary contributing ion currents noted; (b) enhanced automaticity predominantly decreases the duration of phase 4; (c) suppressed automaticity prolongs phase 4; (d) sodium channel blockade slows phase 0 depolarization and thus slows conduction rate; (e) pharmacologically uncovered Brugada syndrome involves a difference in depolarization and repolarization characteristics between the epicardium and endocardium, leading to spontaneous depolarizations during phase 2; (f) transmural dispersion of repolarization manifesting on the ECG as QT prolongation can lead to early afterdepolarizations during phase 3 and torsade de pointes; (g) calcium overload in the cytosol, predominantly as a result of digoxin toxicity, leads to spontaneous depolarizations during phase 4. Abbreviations: I Ca-L L-type calcium channel current, I Ca-T T-type calcium channel current, I f hyperpolarizing sodium current, I K1 inward rectifier potassium channel, I Kr rapid delayed rectifier potassium current, I Ks slow delayed rectifier potassium current, I Na fast inward sodium current, I to transient outward potassium current, I K1 inward rectifier potassium channel, I Kr rapid delayed rectifier potassium current, I Ks slow delayed rectifier potassium current, I Na fast inward sodium current, I Na/Ca sodium/calcium exchanger, I to transient outward potassium current, Na/K ATPase sodium/potassium adenosine triphosphatase
Enhanced automaticity, affecting phase 4 (supraventricular tachycardias, atrial fibrillation, APCs, VPCs, and ventricular arrhythmias)
Suppressed automaticity, affecting phase 4 (bradyarrhythmias)
Blocked or slowed conduction in phase 0 (bradyarrhythmias and tachyarrhythmias with widening of the QRS)
Changes in depolarization and repolarization between the epicardium and endocardium causing arrhythmias in phase 2 (Brugada syndrome)
Altered repolarization, resulting in transmural dispersion of repolarization in phase 3 triggering early afterdepolarizations (prolonged QTc, torsade des pointes, and VF) (Chap. 49)
Altered calcium cycling resulting in intracellular calcium overload, triggering delayed afterdepolarizations in phase 4 (atrial fibrillation, ventricular arrhythmias, and digoxin-specific arrhythmias)
48.3 Specific Proarrhythmic Mechanisms Illustrated
48.3.1 Enhanced Automaticity
Automaticity may be enhanced by increased sympathetic stimulation or parasympathetic withdrawal. This can allow an ectopic focus to become the dominant source of the heart rhythm. The result is sinus tachycardia if the sinoatrial (SA) node is stimulated the most; atrial tachycardia or premature atrial contractions if atrial tissue is affected more; accelerated junctional rhythm if the atrioventricular (AV) node’s rate of excitability exceeds the sinoatrial node rate; and ventricular tachycardia, premature ventricular contractions, or accelerated idioventricular rhythm if the ventricle is the predominant site of enhanced automaticity.
Because of the complex regulatory mechanisms affecting heart rate and contractility, there are multiple ways that drugs can increase automaticity by acting through β-adrenergic receptors, L-type calcium channels, cyclic adenosine monophosphate (cAMP), sodium channels, potassium channels, and adenosine receptors. Cytosol calcium concentration is vitally related to sinus node rate, myocardial contractility, and excitability. The cytosolic and extracellular calcium concentrations are modulated through the sodium/calcium exchanger and the sodium/potassium ATPase. Cytosolic calcium is also cycled between the sarcoplasmic reticulum via the ryanodine receptor, and this is modulated by calmodulin, calsequestrin, and the sarcoplasmic reticulum energy-requiring calcium pump (SERCA) (Chap. 4). The cytosolic phosphorylation and activation of these calcium-cycling proteins not only is regulated by cAMP (generated by the G-protein coupled β-adrenergic receptor) but also by the phosphatidylinositol 3-kinase pathway activated by insulin.
Because of the complexity of the pathways involved, many different types of drugs have a similar impact in contractility. Sympathomimetics increase intracellular cAMP by activating the Gs-coupled beta adrenergic receptor (Chap. 5). Milrinone as a phosphodiesterase inhibitor prevents the breakdown of cAMP. Calcium and digoxin both increase calcium in the cytosol, increasing both contractility and the potential for excitability. Methylxanthines such as caffeine inactivate the inhibitory effect of adenosine on the adenosine-sensitive potassium channels (IKado) in the atria and the adenosine A1 receptors that are also coupled to cAMP. Atropine counteracts the inhibitory effect of the parasympathetic nervous system by blocking muscarinic M2 receptors.
Most arrhythmias that result from these drugs are benign and self-limited. These include sinus tachycardias and atrial tachycardias, common in patients with pulmonary diseases not only on account of the interrelation between the heart and lungs but also because of the frequent requirement for beta-agonists and methylxanthines [3, 4]. Right ventricular outflow tract ventricular tachycardia is triggered by catecholamines and cAMP and may also occur in response to beta agonists.
To counteract the effects of these drugs, particularly in overdose situations, it is more effective to address the dysrhythmias by inhibiting a different part of the regulatory cycle than the one currently affected by the medication. Therefore, in tachycardias from beta-agonists, non-dihydropyridine calcium channel blockers are useful, as well as adenosine for acute dysrhythmias. The dihydropyridine calcium channel blockers act primarily peripherally resulting in a reflex sympathetic activation that counteracts any inhibition of cardiac cells; therefore, dihydropyridine calcium channel blockers would not be useful in patients with excessive sympathetic stimulation [5]. Beta-blockers are helpful in treating tachycardias resulting from thyroid hormone derivatives and methylxanthine overdose, but in cocaine intoxication, they can cause hypertensive crisis by leaving the patient with unopposed alpha receptor activation. Cocaine and other sympathomimetics that act centrally as well as peripherally are best initially treated with centrally acting drugs such as benzodiazepines. Peripherally acting sympathomimetics such as dopamine and dobutamine are short acting, but since they are typically used in hemodynamically compromised patients, it is often necessary to continue these drugs and use amiodarone to suppress arrhythmias. Enhanced automaticity is illustrated in Figs. 48.2 and 48.3.
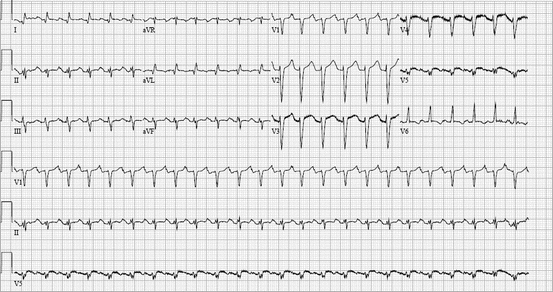
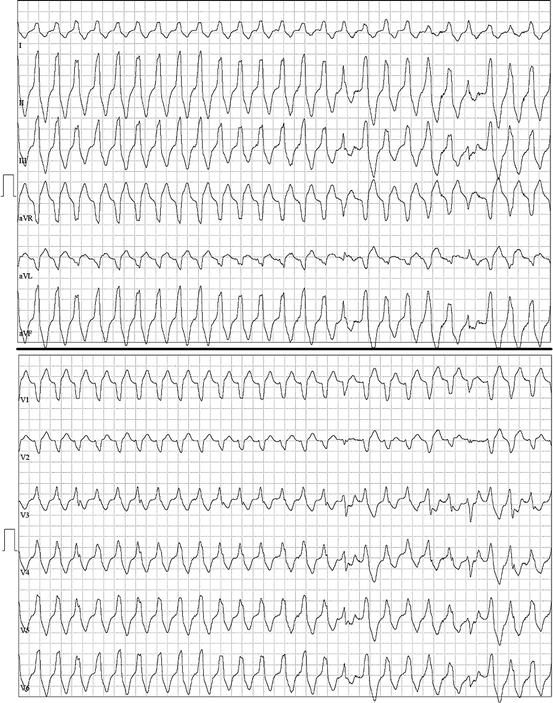
Fig. 48.2
A 60-year-old man with no prior history of arrhythmias, admitted with acute decompensated heart failure, was initiated on a beta-agonist, dobutamine. Although his hemodynamics improved, he had frequent PACs which stimulated runs of a narrow-complex tachycardia that terminated with adenosine. These episodes subsided with discontinuation of the beta-agonist
Fig. 48.3
A 59-year-old man with no prior history of arrhythmias presented for preoperative stress testing. During treadmill stress testing, he developed frequent ectopy and monomorphic ventricular tachycardia, thought to be an outflow tract ventricular tachycardia. The tachycardia has features of a catecholamine-driven ventricular tachycardia that was entirely suppressed by beta-blockade
48.3.2 Suppressed Automaticity
In patients receiving beta-adrenergic antagonists (class II antiarrhythmics) or calcium channel blockers (class IV antiarrhythmics), the calcium current causing the slow action potential is suppressed in the sinoatrial and atrioventricular nodes. Patients receiving high doses of these drugs frequently demonstrate sinus bradycardia or AV blocks, most commonly first-degree or second-degree Mobitz type 1 [6]. Automaticity may also be suppressed by increasing parasympathetic tone, such as with reversible acetylcholinesterase inhibitors such as donepezil and galantamine, which are commonly found to cause bradycardia (Chap. 6). Clonidine centrally inhibits the sympathetic nervous system and increases the parasympathetic tone as an alpha-2 agonist and is also associated with bradyarrhythmias (Chap. 39). Figures 48.4 and 48.5 demonstrate a pharmacologic suppression of automaticity.
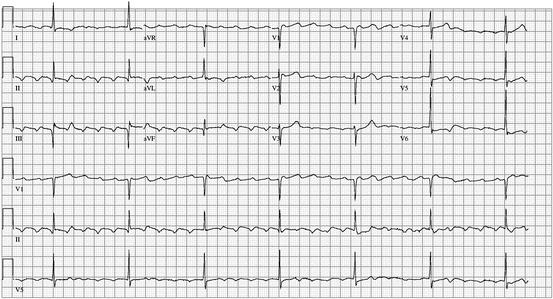
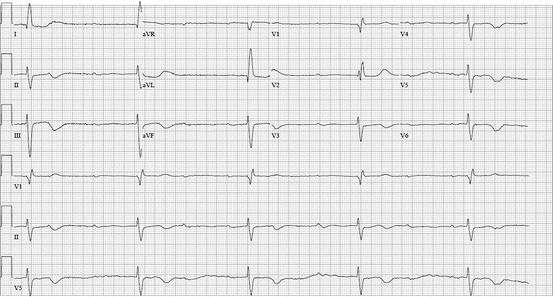
Fig. 48.4
An 80-year-old man with chronic atrial flutter accidentally took two diltiazem extended release tablets instead of his usual single 360-mg daily tablet. He was admitted with lightheadedness and atrial flutter with a ventricular rate of 30–40 beats per minute. His symptoms and bradycardia resolved with treatment with calcium, time, and bed rest
Fig. 48.5
An 82-year-old patient developed acute renal insufficiency while continuing her maintenance atenolol. She had symptomatic bradycardia with hypotension that responded to boluses of glucagon. She ultimately required dual-chamber permanent pacemaker placement as she continued to have intermittent AV block
Arrhythmias increase in severity as the amount of calcium channel blocker or beta-blocker ingested increases. An investigation of poison control records demonstrated that unintentional ingestion of small doses of additional medication is frequently asymptomatic [7]. Most asymptomatic patients can be managed as an outpatient if they are not markedly bradycardic. Patients that ingest more than one cardiac drug have the greatest risk of morbidity and mortality from beta-blocker overdose [8]. Calcium channel blocker and beta-blocker overdose together are particularly problematic, because not only do they profoundly slow the heart rate, they also can profoundly suppress contractility, causing cardiogenic shock. Both aspects must be aggressively addressed to effectively treat calcium channel blocker or beta-blocker overdose. Temporary pacing alone does not improve contractility. Therefore, pharmacologic intervention to counteract the effect of the calcium channel blocker or beta-blocker is required.
Calcium infusion is a logical treatment for calcium channel blocker overdose that also benefits patients who overdose on beta-blockers. Interestingly, it leads to improvements only in the mechanical contractility and not the electrical abnormalities [9]. Glucagon is a hormone secreted by the pancreas to stimulate gluconeogenesis in times of hypoglycemia; it acts through a different receptor coupled to the Gs protein that activates protein kinase A to increase cAMP in the cells independent of the beta-adrenergic receptor. It is effective for calcium channel blocker and beta-blocker overdose, and a clinical effect is rapidly seen with its use. It primarily results in an increased heart rate and increased cardiac output and has little effect on blood pressure. It frequently causes GI distress, however, and tachyphylaxis prevents its long-term use. There have been no randomized studies of glucagon use in humans [10].
As noted above, calcium cycling in the cardiac myocytes is also regulated by phosphatidylinositol-3 kinase, which is activated by insulin and insulin-like growth factor. Using this pathway, insulin is an effective treatment for cardiogenic shock from calcium channel blocker and beta-blocker overdose. Insulin is not proarrhythmic and can allow rapid weaning of pressors by reversing the cardiogenic shock caused by the drugs [11]. Insulin is administered with sufficient glucose to prevent hypoglycemia and potassium to prevent hypokalemia and thus is called a “hyperinsulinemia/euglycemia” protocol [12]. Additionally, insulin and glucose ensure that nutrients are supplied to the myocardium. Hyperinsulinemia/euglycemia treatment is not associated with tachyphylaxis like glucagon is, and as long as patients are closely monitored, it is associated with a low risk of adverse effects [12]. Studies in pig models of beta-blocker toxicity actually show significantly better response with better survival when treated with hyperinsulinemia/euglycemia therapy compared to vasopressors such as epinephrine and vasopressin [13]. Clinically, calcium channel blocker and beta-blocker toxicity range from patients taking normal doses of their prescribed medication who develop some interfering disturbance, such as a patient on atenolol who develops renal failure, to massive ingestions as suicidal attempts. Specific therapy with calcium, glucagon, or hyperinsulinemia/euglycemia is required only if patients develop cardiogenic shock. In medically refractory cases of shock, mechanical hemodynamic support may be required. Figures 48.4 and 48.5 demonstrate significant changes in rhythm precipitated by small changes in medication dose (Fig. 48.4) or renal clearance (Fig. 48.5).
48.3.3 Slowing of Phase 0 Depolarization
The primary effect of sodium channel blockers is to slow the initial phase 0 depolarization of the cardiomyocyte. Because these fast sodium channels are not present in the sinoatrial node and atrioventricular node, these drugs have minimal effect on the sinus rate. By slowing phase 0, each myocyte takes longer to depolarize. At high levels of sodium channel blockade, the QRS on the surface ECG can prolong noticeably. Sodium channel blockers, particularly those in Vaughan Williams classes IA (quinidine, procainamide, and disopyramide) and IC (flecainide and propafenone), are felt to be toxic if the patient’s QRS width has increased by >25 %. However, these drugs are proarrhythmic at any dose and should be used with caution. For example, patients on quinidine for atrial fibrillation had a higher mortality than patients with atrial fibrillation who did not take quinidine, without any notable QRS widening [14]. Additionally, class IC antiarrhythmics were associated with increased mortality in patients with prior myocardial infarction in the Cardiac Arrhythmia Suppression Trials, despite monitoring for drug toxicity [15–17].
Class IA and class IC antiarrhythmics bind to sodium channels in their active state. The more frequently the sodium channels are in their active state, the more effect the antiarrhythmic has on the channels. This property is called the use dependence phenomenon. Consequently, at faster heart rates, the antiarrhythmic will have more effect, and the QRS will be wider. Thus, sometimes, exercise ECGs can be performed to monitor for excessive sodium channel blocker effect, to evaluate prolongation of the QRS with faster heart rates. Use dependence is illustrated in Fig. 48.6. Because of extreme QRS widening from use dependence, differentiating ventricular tachycardia from supraventricular tachycardia with aberrant conduction may be difficult, if not impossible, in sodium channel blocker toxicity states, as shown in Fig. 48.7. The algorithms used to differentiate ventricular tachycardia from supraventricular tachycardia on surface ECG were all developed with patients who were on potentially confounding antiarrhythmics excluded. Therefore, there is no validated approach in these patients to diagnose VT.
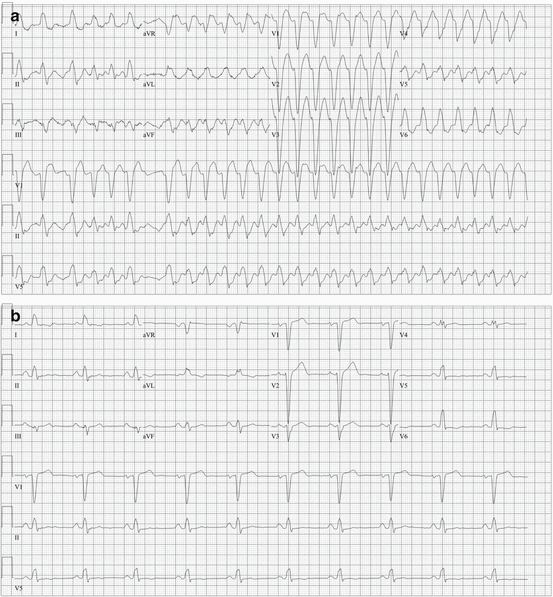
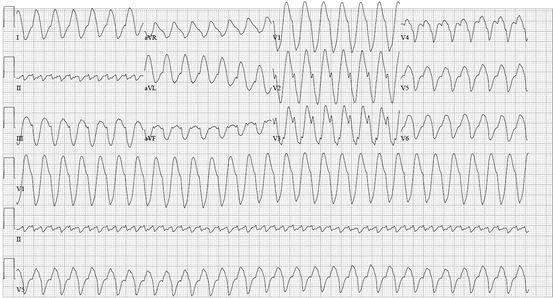
Fig. 48.6
A 78-year-old woman with atrial fibrillation and atrial flutter treated with flecainide and metoprolol presented with palpitations and a wide-complex irregular tachycardia (a). She underwent cardioversion as her underlying rhythm was determined to be atrial flutter with rapid, variable ventricular conduction. With a slower heart rate, her QRS became significantly narrower (b)
Fig. 48.7
Rapid tachyarrhythmias combined with the use dependency of flecainide can cause arrhythmias that are very difficult to differentiate between a supraventricular tachycardia with aberrancy and ventricular tachycardia. A 64-year-old man with paroxysmal atrial fibrillation treated with “pill-in-the-pocket” flecainide presented with chest pain and hemodynamic instability. He later reported that he took flecainide “whenever he felt bad” and had taken a total of 30 50-mg flecainide tablets over 4 days. His initial ECG meets published criteria for VT, particularly with regard to the width of the QRS complex; however, published criteria exclude QRS widening due to antiarrhythmic drugs. Lead II resembles typical atrial flutter, and it is impossible to differentiate with certainty whether this arrhythmia is ventricular tachycardia or 2:1 atrial flutter with marked QRS widening due to the use dependence phenomenon characterizing flecainide. He was cardioverted and treated with intravenous sodium bicarbonate. The following day, his QRS width had normalized
The safest approach is to assume that whatever the rhythm is, in the setting of antiarrhythmic toxicity, it is potentially more unstable than it would be otherwise. Therefore, even in hemodynamically stable patients, urgent sedation followed by synchronized direct current cardioversion is the treatment of choice. Beta-blockade will help with preventing tachycardia and blocking catecholamines, preventing to a large degree the recurrence of proarrhythmia [18]. Theoretically, a different sodium channel blocker, lidocaine, could be used for VT in this situation, but it is preferential to avoid having multiple concurrent antiarrhythmics if possible, to minimize proarrhythmia. Intravenous sodium bicarbonate is useful in overcoming the sodium blockade both by raising the sodium concentration slightly and raising the pH slightly to reduce the sodium channel blocker’s affinity for the sodium channel. Sodium bicarbonate actually prolongs urinary excretion of sodium channel blockers such as flecainide, but in toxicity situations, it is more important to disrupt the affinity between the flecainide and the sodium channel than to expedite its excretion from the body. A similar strategy is used in treating tricyclic antidepressant toxicity.
48.3.4 Atrial Flutter with 1:1 Conduction
Atrial fibrillation and atrial flutter are frequently alternating arrhythmias within the same patient (see also Chap. 50). Patients with atrial flutter have a >50 % risk of developing atrial fibrillation in 10 years [19], and patients with atrial fibrillation have a 30 % chance of developing atrial flutter. The development of atrial flutter becomes increasingly likely with treatment with a sodium channel blocker, which can organize the atrial arrhythmia into a macroreentrant circuit. Furthermore, the sodium channel blockade slows down the flutter cycle length from the typical 200–250 ms of typical flutter to a cycle length of 250–300 ms. Without the effects of antiarrhythmic therapy, the AV node only rarely can conduct atrial flutter faster than a 2:1 atrioventricular ratio. With sodium channel blockade, atrial flutter at slower atrial cycle length can conduct at a 1:1 ratio. This paradoxically increases the ventricular rate in patients with atrial flutter on sodium channel blockers, to the point of impending hemodynamic collapse.
One study of eight patients with 1:1 atrial flutter noted that the cycle length conducted 1:1 during flutter was significantly shorter than the shortest cycle length achievable when pacing patients during electrophysiology study [20]. This finding indicates that enhanced conduction across the AV node, most likely caused by sympathetic stimulation with catecholamines, enhances the conduction across the AV node. Despite its acute presentation, patients who develop 1:1 atrial flutter do not have a significantly worse prognosis compared to patients who do not develop this arrhythmia [21]. Therefore, sodium channel blockers should be combined with a chronic AV node-blocking medication such as a beta-blocker, diltiazem, verapamil, or digoxin. Despite this practice, 1:1 atrial flutter sometimes does occur in the setting of chronic therapy with AV node-blocking drugs. Because 1:1 atrial flutter can occur at any dose of sodium channel-blocking medication, it does not definitively represent toxicity. It should be treated with AV node-blocking agents and synchronized cardioversion if the rhythm persists. Ultimately, atrial flutter ablation is indicated as a highly effective treatment for atrial flutter. Atrial flutter with 1:1 atrioventricular conduction is illustrated in Fig. 48.8.
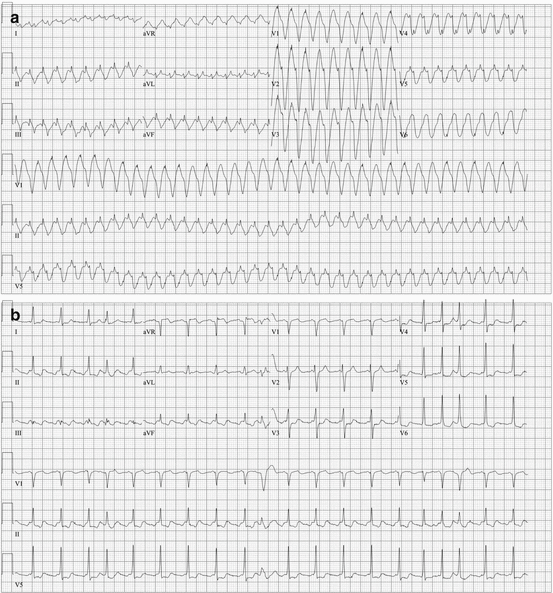
Fig. 48.8
A 60-year-old female with a history of atrial flutter was started on chronic metoprolol and “pill-in-the-pocket” flecainide. A subsequent physician discontinued the chronic metoprolol due to side effects. Later, when her palpitations recurred, she took flecainide without the metoprolol. Since she felt worse, she presented to the emergency department, where the following ECGs were obtained within 2 min of each other. Between ECG (a) and ECG (b), all the patient received was a single dose of benzodiazepine for sedation in preparation for cardioversion. Note that the R–R interval in ECG (a) is exactly the same as the flutter cycle length in ECG, 270 ms (b), proving that the wide complex tachycardia on ECG (a) is indeed atrial flutter with 1:1 atrioventricular conduction
48.3.5 Drug-Induced Brugada Syndrome
The SCN5A sodium channel is defective in Brugada syndrome, a genetic condition that confers an increased risk of sudden cardiac death. The ECG in a patient with Brugada syndrome may have a normal ECG at baseline, and the pattern can be brought out by administering a sodium channel blocker (Fig. 48.9). Arrhythmias occur when abnormal depolarization and repolarization of the epicardium develop, allowing spontaneous depolarization of the epicardium during phase 2 of the cardiac cycle, sometimes called phase 2 reentry [22]. In fact, patients with a pharmacologically inducible Brugada type 1 pattern on their ECG are at a higher risk of ventricular arrhythmias [23]. ICD implantation is indicated in any patient with syncope, ventricular arrhythmias, or aborted cardiac arrest who has a pharmacologically inducible Brugada type 1 pattern on ECG (Chap. 53). On the other hand, a patient with a pharmacologically induced Brugada type 1 pattern with no symptoms concerning for malignant arrhythmias may be at an increased risk of ventricular arrhythmias [24], but management and risk stratification in this patient population is controversial. The management of a patient with an incidental finding of a Brugada pattern ECG in the context of sodium channel-blocking drugs predominantly involves discontinuation of the sodium channel blocker and education of the patient to avoid similar drugs (see www.brugadadrugs.org) [25, 26]. These drugs are listed in Table 48.1 . The management of patients with electrical storm who have a medication-induced Brugada pattern is similar to those with Brugada syndrome [27].
Table 48.1
Drugs that can cause drug-induced Brugada pattern are organized by pharmacologic category [22]
Medication class | Examples |
|---|---|
Antiarrhythmics, class IA | Procainamide, disopyramide, cibenzoline |
Antiarrhythmics, class IC < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|