Principles of Inheritance and Genetics of Congenital Heart Disease
“Pick a rare disease and study it”
Principles of Inheritance of Congenital Heart Disease
Multifactorial Inheritance
Most forms of congenital heart disease (CHD) have a multifactorial etiology. This means that the development of a congenital heart malformation is determined by the interaction of several environmental and genetic factors. Although this may seem like a rather vague and imprecise mechanism, there are several principles of the multifactorial model that are borne out by empiric observations. These principles are useful for purposes of genetic counseling.
In the multifactorial model, the risk of occurrence, in general, is related to the frequency with which the defect occurs in the general population. For example, if one considers all CHDs, the frequency of their occurrence in the general population is approximately 0.7 of 100 live births. Therefore, if no members of a family have CHDs, the risk that a couple will have a child with CHD is approximately 0.7%.
The risk of an individual being born with CHD is increased if a first-degree relative (a parent or a sibling) has CHD. The risk is about 2% to 5%, assuming a multifactorial inheritance pattern. If it is a relatively common CHD such as ventricular septal defect (VSD), the recurrence risk will be at the higher end of this range, whereas if it is a relatively uncommon defect such as tricuspid atresia, it will be at the lower end of this range. The recurrence risk of CHD, if a first-degree relative has CHD, will depend on the specific defect and whether there is a gender predilection for the specific cardiac defect.
Much of the data on recurrence risk is based on empiric observational studies, and significant differences exist among different studies because of the manner in which the studies were performed (see Fig. 8-1). For example, an early study of the recurrence risk of aortic stenosis in children born to mothers with aortic stenosis suggested a recurrence risk as high as 26%. However, this study likely included significant ascertainment bias. In addition, the diagnosis of aortic stenosis in the children was based on physical examination only. The preponderance of the studies, however, suggests that the recurrence risk for aortic stenosis, pulmonary stenosis, and VSD in children of a mother or father with one of these defects is approximately 5% (see Table 8.1). In the second natural history study (NHS-2), there was minimal, if any, ascertainment bias, but the presence of CHD in the children was established
on the basis of history alone, and the recurrence rate was <5%. There have been several studies of atrioventricular septal defect (AVSD) suggesting that the recurrence risk is greater than the 5% that has been observed for aortic stenosis, pulmonary stenosis, and VSD.
on the basis of history alone, and the recurrence rate was <5%. There have been several studies of atrioventricular septal defect (AVSD) suggesting that the recurrence risk is greater than the 5% that has been observed for aortic stenosis, pulmonary stenosis, and VSD.
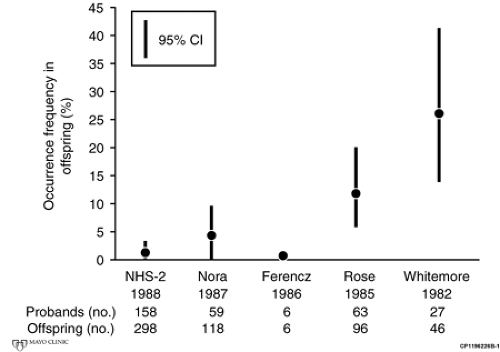 Figure 8.1 • Comparison of the results of several studies of the recurrence risk for aortic valve stenosis. CI, confidence interval. See Driscoll D, Michels V, Gersony W, et al. Occurrence risk for congenital heart defects in children of patients with aortic stenosis, pulmonary stenosis, or ventricular septal defect: Report of the second natural history study of congenital heart defects. Circulation. 1993;87:I114–I120 . |
Theoretically, if a CHD is more common in one gender than in the other, the risk of recurrence in the child will be higher if the defect is in the parent for whom the risk of incidence is lower. For example, aortic stenosis is more common in men than in women. Presumably, in a multifactorial model, if a woman has aortic stenosis, a greater load of
environmental–genetic factors exists to cause the defect in the woman than in the man. This greater “environmental–genetic load” should increase the chances of the defect recurring in the child of that mother. However, empiric studies do not bear this out in all cases. It is not perfectly clear why the multifactorial model breaks down in this situation. It could probably be because not all cases of aortic stenosis, or for that matter any form of CHD, are explained by the multifactorial model. An important number of cases may be single gene defects, examples of the Knudson double hit hypothesis, a mixed model of multifactorial and single gene defects, or somatic gene mutations. Furthermore, empiric risk data have not been studied for all categories of recurrence risks relative to gender.
environmental–genetic factors exists to cause the defect in the woman than in the man. This greater “environmental–genetic load” should increase the chances of the defect recurring in the child of that mother. However, empiric studies do not bear this out in all cases. It is not perfectly clear why the multifactorial model breaks down in this situation. It could probably be because not all cases of aortic stenosis, or for that matter any form of CHD, are explained by the multifactorial model. An important number of cases may be single gene defects, examples of the Knudson double hit hypothesis, a mixed model of multifactorial and single gene defects, or somatic gene mutations. Furthermore, empiric risk data have not been studied for all categories of recurrence risks relative to gender.
TABLE 8.1 Recurrence Rates for Aortic Stenosis, Pulmonary Stenosis, and Ventricular Septal Defect from the Second Natural History Study of Congenital Heart Disease | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||
Single Gene Defects
Over the last 15 years, an increasing number of single gene defects have been discovered to be associated with isolated CHDs. Examples include supravalvar aortic stenosis (elastin gene), atrial septal defect (ASD) (genes that encode transcription factors TBX5, NKX 2.5, and GATA4, and the structural protein MYH6), hypertrophic cardiomyopathy (mutations in multiple genes that encode proteins of the cardiac sarcomere), and tetralogy of Fallot, as well as related defects (JAG, which is allelic to Alagille syndrome). If the CHD results from a single gene defect, recurrence risks would reflect a Mendelian pattern such as autosomal dominant or recessive. It is important to recognize these situations because the recurrence risks will be much greater than those if the inheritance pattern is multifactorial. Variable expression and incomplete penetrance need to be considered.
In addition to hereditary germ line mutations, somatic mutations in genes that relate to cardiac development have been demonstrated only in the heart tissue in some cases of sporadic CHD.
Marfan Syndrome
Marfan syndrome is an autosomal dominant condition with variable expression that results from a mutation in the gene that encodes for fibrillin-1. Less commonly, it is due to a mutation in transforming growth factor β (TGF β). It is characterized by tall stature, pectus excavatum or carinatum, reduced upper to lower body segment ratio (approximately <0.88, but it is age dependent), an arm span to height ratio >1.05, long thin fingers (arachnodactyly), pes planus, lax joints, long thin face, highly arched palate, myopia, dislocated lenses, skin stria, spontaneous pneumothorax, dural ectasia, mitral valve prolapse, aortic aneurysm, and aortic dissection. A strict clinical criteria (Ghent Criteria, see Table 8.2) must be used to make the diagnosis. Although assays for fibrillin-1 and TGF β are available, the results of these tests must be interpreted carefully because patients without identifiable gene defects can still have the features, risks, and complications of Marfan syndrome. Furthermore, gene variations of uncertain clinical significance may be detected. Therefore, it still is important to make a clinical diagnosis. The criteria listed in Table 8.3 are a useful guide in determining who likely has and who does not have Marfan syndrome.
The major cardiac issues in Marfan syndrome are aortic aneurysm and aortic dissection, aortic valve insufficiency, and mitral valve prolapse and insufficiency. The leading causes of death in Marfan syndrome are aortic rupture and aortic dissection. Hence, identification of patients with Marfan syndrome is important so that these problems can be diagnosed early and treated appropriately. There is clear evidence that treatment of patients with Marfan syndrome with β-blocking drugs slows the progression of aortic root dilation and reduces the risk of aortic rupture and dissection. There is general agreement that all patients with
Marfan syndrome and aortic root or sinus of Valsalva dilatation should be treated with β-blockers unless there are comorbid contraindications. For patients who cannot be treated with β-blockers, it seems reasonable (but there are only limited studies) to use other agents to reduce blood pressure. Whether patients with Marfan syndrome without aortic dilation should be treated with β-blockers is controversial. Because aortic dilation is a progressive process, imaging of the aorta, at least annually, is necessary. More frequent imaging will be necessary in some patients. Because Marfan syndrome does not involve just the ascending aorta, it is important to periodically image the entire aorta.
Marfan syndrome and aortic root or sinus of Valsalva dilatation should be treated with β-blockers unless there are comorbid contraindications. For patients who cannot be treated with β-blockers, it seems reasonable (but there are only limited studies) to use other agents to reduce blood pressure. Whether patients with Marfan syndrome without aortic dilation should be treated with β-blockers is controversial. Because aortic dilation is a progressive process, imaging of the aorta, at least annually, is necessary. More frequent imaging will be necessary in some patients. Because Marfan syndrome does not involve just the ascending aorta, it is important to periodically image the entire aorta.
TABLE 8.2 Diagnostic Criteria for Marfan Syndrome | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


