Prevention of Sudden Death in Heart Failure
Philip B. Adamson
Emilio Vanoli
Sudden cardiac death is defined as death within minutes to hours from the onset of symptoms in a patient who otherwise was clinically stable (1,2). This definition seems simple but, in reality, determining the cause of sudden cardiovascular collapse can be challenging (3). Clinical trials differ about which deaths are considered “sudden,” making accurate comparison across trials difficult (4). For example, the Cardiac Insufficiency and Bisoprolol Trial (CIBIS) classified unwitnessed deaths that occurred during sleep as “unknown cause” rather than “sudden,” whereas other trials consider overnight deaths as “sudden” (5). Furthermore, the true epidemiology of sudden cardiac death is difficult to quantify, but estimates in the U.S. population range from 300,000 to 500,000 annual deaths that are attributable to sudden cardiac arrest (6,7,8,9,10).
Even with ambiguity of numbers, sudden death (SD) is a major cause of premature mortality in patients who otherwise have stable, even minimally, symptomatic cardiovascular disease. The cardiology community’s frustration with SD was clearly stated by Prof. Bernard Lown, who in 1979 considered SD as “the most important challenge facing modern cardiology” (11). Despite significant efforts examining the mechanisms of lethal cardiac arrhythmias over the past 30 years, the syndrome of sudden cardiac death still remains an important challenge facing modern cardiology (12,13). This chapter will focus on the epidemiology of SD in order to identify at-risk patient groups, and will organize available evidence supporting the rationale of pharmacologic arrhythmia suppression and immediate termination strategies when lethal arrhythmias develop. These data will be summarized in a practical approach to provide the currently accepted standard of care for SD prevention in patients with chronic heart failure.
Clinical Characteristics of Sudden Death
Most sudden cardiac deaths result from ventricular tachyarrhythmias, while bradyarrhythmias, great vessel rupture, or pulmonary embolism account for other etiologies of sudden cardiovascular collapse (14,15,16,17,18,19,20,21,22,23,24) (Fig. 41-1). The overwhelming majority of people who develop sustained ventricular tachyarrhythmias outside the hospital die, since immediate electrical therapy is required for any hopes of survival (25,26). For every minute a ventricular arrhythmia is established, the chance of survival is reduced by 10%, so that 10 minutes after cardiovascular collapse, there is virtually no chance of survival (25). The need for immediate therapy helps explain why less than 5% of patients in the United States who have out-of-hospital cardiac arrest survive, even in areas of well-developed rapid response systems. An aspect of SD that is critical to understanding the syndrome is that, by definition, it is not preceded by warning events, even in patients with symptomatic heart failure. A post hoc analysis of the CIBIS-II trial demonstrated that only 20% of the patients who died suddenly during follow-up had worsening heart failure symptoms or warning hospitalization prior to the lethal event (27). In stark contrast, worsening heart failure symptoms and hospitalization preceded almost 90% of the patients who died of pump failure (27). Therefore, since warning symptoms are not present to identify patients at risk for impending sudden cardiovascular collapse, primary prevention of SD must rely on identifying high-risk patients before their fatal event.
Sudden cardiac death generally occurs in the presence of some form of structural heart disease (28,29,30,31,32), such as
myocardial ischemia or infarction (33,34,35), dilated cardiomyopathy, hypertrophic cardiomyopathy (36), or genetically determined primary electrophysiological abnormalities such as long-QT syndrome (37) or Brugada syndrome (38). Of all the possible associated diseases, ischemic heart disease is the most common underlying pathology that renders the ventricle electrically unstable (33,34,35,39,40). Thus, SD may occur in profoundly different clinical settings (Fig. 41-2).
myocardial ischemia or infarction (33,34,35), dilated cardiomyopathy, hypertrophic cardiomyopathy (36), or genetically determined primary electrophysiological abnormalities such as long-QT syndrome (37) or Brugada syndrome (38). Of all the possible associated diseases, ischemic heart disease is the most common underlying pathology that renders the ventricle electrically unstable (33,34,35,39,40). Thus, SD may occur in profoundly different clinical settings (Fig. 41-2).
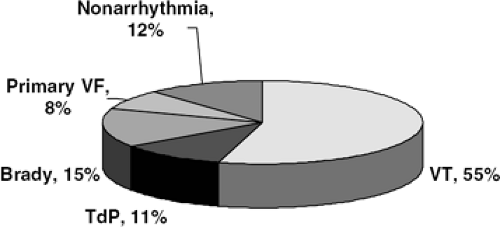 Figure 41-1 Causes of sudden cardiovascular collapse. TdP, torsade du pointes; VT, ventricular tachycardia; VF, ventricular fibrillation; Brady, bradycardia. Nonarrhythmic causes of sudden death include pulmonary embolism, great vessel rupture, and lethal stroke. (Adapted from Bayes de Luna A, Coumel P, Leclercq JF. Ambulatory sudden cardiac death. Mechanisms of production of fatal arrhythmia on the basis of data from 157 cases. Am Heart J. 1989;117:151–159 .) |
The most difficult patients to identify are those who die from lethal arrhythmias as the first manifestation of ischemic heart disease. In contrast to an overall trend toward a reduction in cardiac mortality (10,41), the incidence of sudden arrhythmic death in subjects free of overt ischemic heart disease has remained unaltered over the last 20 years (10,23,32,33). Annually, more than 50,000 Americans die of unheralded SD that is often the first, and last, manifestation of ischemic heart disease. This major public health issue has received little attention, presumably due to the general belief that high-risk individuals cannot be identified with enough predictive accuracy to warrant specific preventive interventions. Indeed, these individuals are usually not under the care of a physician and the unexpected nature of their death precludes any prediction or preparedness.
Autopsy series performed in individuals who died suddenly without prodromal symptoms usually find the presence of significant obstructive coronary artery disease with evidence for a very recent thrombotic event (33). These individuals apparently respond to their first ischemic episode with a lethal arrhythmia, since most autopsy reports fail to actually demonstrate the presence of acute myocardial necrosis, implying that the cardiac arrest occurred immediately in response to the ischemic event. Inherited traits produce familial trends in both risk for SD and autonomic dysfunction that contribute to the overall risk profile of individuals who die suddenly and unexpectedly (21,40).
Those few patients who survive unexpected cardiac arrest without a prior history of cardiovascular characterized by significant cardiac autonomic control system dysfunction resulting in imbalances favoring strong sympathetic input with relatively weak vagal control (42). These findings are supported from experimental evidence in canines at high risk for ventricular fibrillation during their first episode of myocardial ischemia (43,44,45). Highrisk animals respond to acute ischemia with strong sympathetic activation and weak vagal reflexes, which lead to the development of ventricular fibrillation (VF). In contrast,
those at low risk for lethal arrhythmias respond to the same ischemic insult with vagal activation and relatively weak sympathetic input, and subsequently have no sustained ventricular arrhythmias. These differences in autonomic response to acute myocardial ischemia, even in the absence of structural heart disease (44,45), are important components of the mechanisms responsible for SD throughout the spectrum of chronic ischemic heart disease, from the first manifestation to end-stage heart failure (46).
those at low risk for lethal arrhythmias respond to the same ischemic insult with vagal activation and relatively weak sympathetic input, and subsequently have no sustained ventricular arrhythmias. These differences in autonomic response to acute myocardial ischemia, even in the absence of structural heart disease (44,45), are important components of the mechanisms responsible for SD throughout the spectrum of chronic ischemic heart disease, from the first manifestation to end-stage heart failure (46).
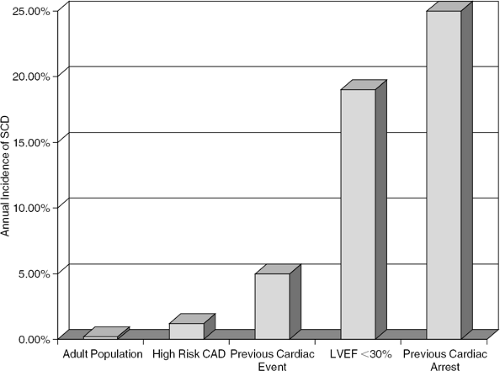 Figure 41-2 Incidence of sudden cardiac death (SCD) in the entire adult population, patients with coronary artery disease (CAD), previous cardiac injury, depressed left ventricular ejection fraction (LVEF), and survivors of cardiac arrest. Note the high incidence of sudden death in patients with left ventricular dysfunction defined as an ejection fraction less than 30%. (Adapted from Larsen MP, Eisenberg MS, Cummins RO, et al. Predicting survival from out-of-hospital cardiac arrest. A graphic model. Ann Emerg Med. 1993;22:1652–1658 .) |
The second general condition in which elevated SD risk can be identified is after ventricular injury by myocardial infarction (MI) (Fig. 41-2). The incidence of post-MI sudden cardiac death has decreased over the past 30 years, however, presumably due to early revascularization therapy that restores circulation to the infarct zone (10,46). The incidence of out-of-hospital sudden cardiac arrest is estimated to be 0.8 per 1,000 subject-years in individuals without clinically recognized heart disease. In patients with overt coronary artery disease (CAD), SD incidence is 13.6 per 1,000 subject-years in subjects with prior MI and 21.9 per 1,000 subject-years in subjects with symptomatic heart failure, illustrating that as heart disease progresses, ventricular electrical stability declines over time (Fig. 41-3) (6). The highest risk for SD following MI appears to be in the first 30 days (47,48), but attempting to reduce this risk by using an implantable cardioverter defibrillator was, at least in one population, associated with increased mortality (49).
Minimization of infarct size after MI with early revascularization strategies (50), coupled with prevention of adverse myocardial and electrical remodeling using angiotensin-converting enzyme (ACE) inhibitors (51,52), aldosterone antagonists (53), and beta-blockers (54) routinely after MI, are effective in reducing the incidence of post-MI SD. Less successful approaches to reduce post-MI SD involved using antiarrhythmic drugs in a primary prevention strategy. Trials using this approach with sodium channel (INa) blockers in post-MI patients from the late 1970s were affected by the change in SD incidence resulting in higher on-treatment mortality compared with placebo, which as a group had an unexpectedly low SD risk (55). Trials investigating antiarrhythmic drug therapy targeting specific ion channels, such as IKr (56) or using amiodarone (57,58) were associated with either increased mortality on treatment (56) or no effect on overall mortality (57). Further refinements in the treatment of acute coronary syndromes, such as platelet inhibition and routine percutaneous catheter-mediated intervention, seemed to impact short-term SD risk, but only assumptions can be made about long-term (years) benefits.
Following MI, autonomic imbalances favoring the sympathetic nervous system can be detected in patients at high
SD risk as measured by autonomic markers such as heart rate variability (HRV) or baroreflex sensitivity (BRS) (59,60,61). These markers have Class I consensus recommendation to aid in post-MI risk stratification with a level of evidence A (62). In patients and animals with a healed MI, quantifying vagal reflex activation by relating blood pressure increases with heart rate slowing (the so-called baroreflex sensitivity test) can stratify longitudinal risk for lethal arrhythmias (59,60). Those at high risk for SD have weaker cardiac vagal reflexes, implying that vagal withdrawal, which probably occurs along with sympathetic activation during progressive cardiac disease, is an important component of the mechanisms involved in lethal arrhythmogenesis. Vagal augmentation by electrical stimulation of the cervical sympathovagal trunk significantly reduces the incidence of ventricular fibrillation (VF) when applied to highrisk animals at the time of vulnerability (63). Interestingly, the ventricular arrhythmia suppressive effect of vagal stimulation was, in large part, still present even when heart rate was maintained by atrial pacing.
SD risk as measured by autonomic markers such as heart rate variability (HRV) or baroreflex sensitivity (BRS) (59,60,61). These markers have Class I consensus recommendation to aid in post-MI risk stratification with a level of evidence A (62). In patients and animals with a healed MI, quantifying vagal reflex activation by relating blood pressure increases with heart rate slowing (the so-called baroreflex sensitivity test) can stratify longitudinal risk for lethal arrhythmias (59,60). Those at high risk for SD have weaker cardiac vagal reflexes, implying that vagal withdrawal, which probably occurs along with sympathetic activation during progressive cardiac disease, is an important component of the mechanisms involved in lethal arrhythmogenesis. Vagal augmentation by electrical stimulation of the cervical sympathovagal trunk significantly reduces the incidence of ventricular fibrillation (VF) when applied to highrisk animals at the time of vulnerability (63). Interestingly, the ventricular arrhythmia suppressive effect of vagal stimulation was, in large part, still present even when heart rate was maintained by atrial pacing.
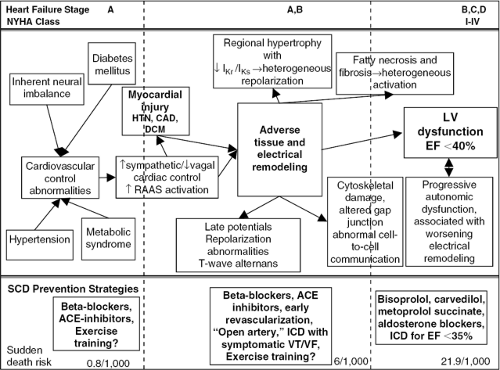 Figure 41-3 An illustration of the proposed mechanisms involved in worsening myocardial and electrophysiologic function as cardiovascular disease progresses from neurohormonal changes associated with risk factors such as hypertension and diabetes, through the development of left ventricular dysfunction. It is important to consider that the outcome of sudden death is the result of tissue and electrophysiologic changes that occur upstream from the clinical arrhythmic event. (Refer to Dinerman JL, Berger R, Haigney MC, et al. Dispersion of ventricular activation and refractoriness in patients with idiopathic dilated cardiomyopathy. Am J Cardiol. 1997;79:970–974 .) |
On the other hand, vagal inhibition by atropine (64) or sympathetic activation by ephedrine (65) also enhances arrhythmic risk and mortality in otherwise low-risk subjects. Patients with conditions such as MI or heart failure, which are characterized by an autonomic control milieu consisting of parasympathetic withdrawal or inherently weak cardiac vagal control coupled with sympathetic activation, have an increased relative risk for lethal arrhythmias.
Patients have an increasing risk for SD as their heart disease progresses from coronary occlusion to heart failure. In this regard, patients with heart failure represent one of the highest-risk groups that are routinely under physicians’ care. It is the intent of this chapter to examine potential mechanisms responsible for risk development and provide a basis for primary and secondary prevention SD prevention strategies in patients with chronic heart failure.
Lethal Arrhythmia Suppression Versus Sudden Death Prevention in Patients with Heart Failure
Heart failure pathophysiology is very complex, with regional differences in fibrosis, hypertrophy, infarction, and ischemia that ultimately result in a global decreased force of contraction coupled with a loss of the normal force-frequency relationship. Multiple mechanisms are involved in the development of left ventricular dysfunction but the intimate relationship between cardiac cell excitation and coupling of that excitation with muscle contraction makes it impossible to isolate tissue, structural, and architectural adverse remodeling from the associated changes in cardiac electrophysiology (65,66,67,68,69,70). The fatal result of these changes (the clinical syndrome of sudden cardiac death) is identified by the presence of lethal arrhythmias (mostly ventricular fibrillation or unstable ventricular tachycardia), but the actual events that lead to arrhythmia development are much more important when considering prevention (Fig. 41-3).
Clinical interventions that effectively reduce the relative risk for SD focus on either the upstream events that eventually lead to the development of lethal arrhythmias, or provide a means for rapid termination of ventricular tachyarrhythmias once they develop. Therefore, SD prevention in patients with chronic heart failure involves two basic mechanisms, each of which will be considered in detail in this chapter.
First, suppressing lethal ventricular tachyarrhythmias can be accomplished by preventing the fixed or functional electrophysiological substrate conducive for arrhythmogenesis and suppressing electrophysiological triggers responsible for initiating a re-entrant circuit. Since the substrate for triggered activity and re-entry is, in large part, dependent on neurohormonal activation in response to injury, it is not surprising that the most effective pharmacologic means to prevent lethal arrhythmias in patients with heart failure is beta-blocker therapy coupled with angiontensin-converting enzyme (ACE) inhibition and aldosterone antagonism. Even with appropriate neurohormonal antagonism, however, the residual risk for SD is still high, which requires inclusion of complementary interventions to effectively reduce overall SD risk.
Along with arrhythmia suppression using appropriate neurohormonal intervention, the second general approach to SD prevention is immediate electrical treatment to terminate potentially lethal ventricular arrhythmias once they are established. This approach is accomplished by use of implantable cardioverter defibrillators designed to deliver either antitachycardic pacing or defibrillation when appropriate arrhythmia sensing occurs. These prevention strategies are synergistic and theoretically produce a >70% relative risk reduction for SD when compared to no intervention (72).
Lethal Arrhythmia Suppression
Cardiac electrophysiology in heart failure patients is the result of complex interactions between local events, such as myocardial ischemia, fibrosis, and replacement of the normal myocardial syncytium, coupled with functional modifications by cardiac control systems primarily arising from the autonomic nervous and renin-angiotensin-aldosterone systems (RAAS). Adverse tissue and architectural remodeling, characteristic of chronic heart failure, is not homogeneous, and this has direct consequences on cardiac electrical properties. In addition to cardiac structural changes, remodeling of cardiac ion channel densities structure, or function, coupled with alterations in cell-to-cell communication, significantly alter both activation and repolarization. This process, called electrical remodeling represents the upstream process by which an arrhythmogenic substrate provides risk for lethal tachyarrhythmias (Fig. 41-3, Table 41-1).
Alterations in Activation and Risk for Sudden Death
Cardiac cell activation and conduction of the action potential through the myocardial syncytium depend on availability of appropriate sodium current and cell-to-cell communication through the proper density and distribution
of gap junctions (73,74,75,76). Adverse remodeling associated with chronic cardiovascular disease and heart failure can alter activation and conduction by influencing sodium channel (INa) densities, especially in areas of myocardial ischemia or infarction (75,76,77,78,79,80,81,82,83). INa function is also altered in heart failure by changes in cytoskeletal support of the channel (77). Conduction abnormalities can arise from altered INa or by functional abnormalities imposed by intracellular fibrosis (82) or changes in cell size (83).
of gap junctions (73,74,75,76). Adverse remodeling associated with chronic cardiovascular disease and heart failure can alter activation and conduction by influencing sodium channel (INa) densities, especially in areas of myocardial ischemia or infarction (75,76,77,78,79,80,81,82,83). INa function is also altered in heart failure by changes in cytoskeletal support of the channel (77). Conduction abnormalities can arise from altered INa or by functional abnormalities imposed by intracellular fibrosis (82) or changes in cell size (83).
Table 41-1 Electrophysiological Consequences of Adverse Tissue Remodeling Leading to a Myocardial Substrate at High Risk for Sudden Cardiac Death | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
These aspects of electrical remodeling can result in a fixed substrate when changes in the ventricular myocardium permanently alter electrophysiological characteristics in the area of change. Examples of fixed electrophysiological abnormalities include fractionated activation across a so-called mottled infarct created by surviving tissue interspersed among necrotic or fibrotic tissue (84,85,86,87,88,89,90). Activation fronts encountering areas of patchy fibrosis or fatty necrosis either alter conduction impedance or allow conduction through the zone by way of surviving tissue that forms conduction channels that traverse the area of patchy necrosis. Impulses conducting through channels can exit from the zone of patchy conduction to encounter excitable tissue and give rise to triggers for re-entrant arrhythmias. This combination of delayed activation, unidirectional block, and discontinuous conduction is thought is be the macroscopic mechanisms responsible for re-entrant tachyarrhythmias.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


