Fig. 28.1
RCT model showing (1) cholesterol efflux, (2) cholesterol esterification by LCAT, and (3) selective hepatic uptake of HDL lipids
One approach has been to increase the quality or plasma concentration of acceptors. Given that phospholipids are the essential cholesterol-binding molecule in HDL and the acyl donor in cholesterol esterification via LCAT, and that apo AI is the LCAT activator, researchers tested various reassembled (r) HDL. One of these was rHDLMilano, which contains phospholipid and apo AIMilano, a variant of apo AI found in an Italian family that was resistant to atherogenesis. Early studies were promising [51], but subsequent tests showed that rHDLMilano infusion remodels the arterial external elastic membrane but does not change lumen dimensions [52]. Infusions of delipidated HDL plasma increased pre-β-like HDL, which is thought to be atheroprotective and reduced α-HDL, the mature form giving a more cardioprotective plasma profile. However, intravascular ultrasound showed only a trend in atheroregression [53]. Other rHDL, apo AI variants, and apo AI mimetics have been tested for atheroregression. Some formulations (CSL-111, CSL-112, and ETC-216) mobilize cellular cholesterol and regress atherosclerotic plaques in animal models, even after only a few treatments [54]. Synthetic peptide analogs of apos also mediate cholesterol efflux [55]. An intrinsic problem with all of these infused agents that remove cholesterol from cells, be they peptides, rHDL, delipidated HDL, or lipid vesicles, is that they lack directionality and would likely remove cholesterol from all tissue sites, including the liver, the target of therapeutic disposal. Therefore, development of agents that selectively enhance macrophage cholesterol efflux will be a challenging task.
There have been various attempts to potentiate the second RCT step, cholesterol esterification via LCAT. This seems to be a sound approach; unlike free cholesterol, which moves reversibly between membrane and lipoproteins, its much more lipophilic ester cannot spontaneously transfer, thereby preventing effluxed cholesterol from transferring back to macrophages. However, the findings in population studies do not support this approach. A common LCAT variant found in patients with the lowest 2 % HDL-C was associated with a 13 % decrease in HDL cholesterol but not with increased risk of MI or other ischemic end points [56]. LCAT activity in CVD cases vs. controls has been compared; these studies revealed that high plasma LCAT activities did not predict reduced CVD risk [57], and other studies showed that plasma LCAT levels had little or no association with future CVD events [58]. Mature HDL is a poor LCAT substrate [59], and in normal subjects, the fraction of HDL cholesterol that is esterified is nearly invariant. Mice overexpressing LCAT have elevated plasma HDL-C but more atherosclerosis [60]. None of these findings support the hypothesis that simply increasing LCAT activity is a viable antiatherogenic therapy.
Studies in genetically altered mice suggest that the terminal RCT step, selective hepatic cholesterol disposal, might be an attractive therapeutic target. Selective uptake is mediated by scavenger receptor class B type I (SR-BI). Mice bearing the SR-BI transgene have reduced plasma HDL-C concentrations but less atherosclerosis, whereas mice in which the SR-BI gene has been ablated have elevated plasma HDL-C concentrations but more atherosclerosis [61–64]. These observations are, of course, the opposite of expectations based on the “high plasma HDL-C concentrations are better” hypothesis. Enhancement of the final RCT step, hepatic cholesterol disposal via SR-BI, is a promising strategy because this step provides RCT directionality.
There are several options. One could alter HDL structure in a way that enhances their hepatic disposal by multiple hepatic receptors. Given that apo E is a ligand for multiple receptors, apo E and its mimetics are attractive vectors for targeting lipoprotein-C for hepatic disposal. Mims et al. synthesized an analog containing all or part of the apo E receptor-binding domain [65]. These analogs also contained a diacyl moiety that irreversibly bound the peptides to lipoprotein surfaces, thereby guiding the lipoprotein to which it was bound to its receptor for endocytosis. Addition of some of these peptides to LDL enhanced its specific uptake and degradation by LDL receptor- and non-receptor-mediated mechanisms. Addition of the peptides to rHDL increased its affinity for the LDL receptor to that of apoE rHDL. Other apo E mimetics have also been tested. One of these, which consists of the receptor-binding region covalently linked to an amphipathic helix, reduces plasma cholesterol levels in mice and rabbits [66, 67]. Another possibility is the reaction catalyzed by streptococcal virulence factor, serum opacity factor (SOF), which disrupts HDL structure, giving among other products a cholesteryl ester-rich microemulsion (CERM) containing nearly all the cholesteryl esters of HDL and only apo E and its heterodimer with apo AII [68]. With its apo E and a high CE content, CERM is a target for hepatic uptake via multiple apo E receptors [69]. Intravenous injection of SOF into wild-type mice reduced plasma cholesterol via hepatic uptake (−43 %) within 3 h; parallel studies with apo E-deficient, and with LDLR-deficient, mice revealed that CERM clearance requires apo E and that most of the hepatic clearance of CERM occurs via the LDLR [70]. However, these approaches are very preliminary and require additional optimization and testing to determine whether they reverse atherosclerosis in mice and other models.
28.3 Plasma TG and Metabolic Syndrome (MetS)
Hypertriglyceridemia, the elevation of plasma TG concentrations, can occur through several mechanisms. Genetic mutations that affect the hydrolysis of plasma TG can lead to severe hypertriglyceridemia. These include a deficiency of lipoprotein lipase (LPL), which hydrolyzes plasma TG associated with chylomicrons and VLDL, or the LPL activator protein, apo CII [71]. Conversely, rare mutations that decrease apo CIII levels are associated with lower TG levels and reduced risk of coronary heart disease [72]. Although isolated severe hypertriglyceridemia is associated with an increased risk of pancreatitis, its link with CVD appears to be associated with other factors that comprise MetS. MetS is defined as the coexistence of three or more of the following: hypertriglyceridemia, a low plasma HDL-C concentration, hypertension, hyperglycemia, and a large waist circumference. However, this cluster of lipid risk factors is also frequently associated with small, dense LDL, low LPL activity, elevated hepatic lipase activity, and hyperinsulinemia, which in association with hyperglycemia produces an insulin-resistant state. The risk factors have been mechanistically linked in a model of systemic steatosis [73], which begins with excess lipolysis within femoral–gluteal fat depots and leads to an excess plasma nonesterified fatty acid concentration (Fig. 28.2; Step 1). As a consequence, the excess fatty acids flow to other sites—to the liver (Step 2) where nonesterified fatty acids are converted to excess TG that is secreted as VLDL (Step 3) producing a hypertriglyceridemic state and to skeletal muscle (Step 4) where it impairs glucose disposal [74, 75] producing hyperglycemia and inducing insulin production followed by hyperinsulinemia. The plasma TG of elevated VLDL are exchanged for the cholesteryl esters of HDL2 and LDL (Step 5) thereby making them TG-rich and cholesterol-poor; [76] this reduces HDL-C. Moreover, the TG-rich HDL and LDL are substrates for hepatic lipase (Step 6), which removes much of their TG and reduces them to their smaller forms, HDL3, and small dense LDL. Finally, nonesterified fatty acids remaining after hepatic and muscle uptake are diverted to central depots (Step 7) resulting in a large waist circumference.
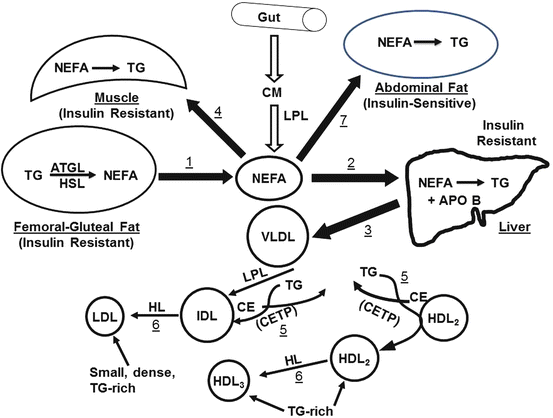
Fig. 28.2
Model of systemic steatosis stemming from excess lipolysis in femoral–gluteal fat depots: ATGL adipose tissue lipase, HL hormone-sensitive lipase, CETP cholesteryl ester transfer protein, CM chylomicrons, IDL intermediate-density lipoproteins, TG triglyceride, NEFA nonesterified fatty acids, LPL lipoprotein lipase. See text for details
According to the model in Fig. 28.2, measures that reduce plasma nonesterified fatty acid concentrations would be expected to relieve MetS. These could include exercise, which reduces fasting and postprandial plasma TG concentrations while increasing HDL, particularly HDL2 [77]. Fibrates are PPARα agonists that are hypotriglyceridemic, an effect that might be mechanistically linked to their in vitro enhancement of nonesterified fatty acid β-oxidation [78], which consumes intracellular nonesterified fatty acids, thereby diverting them from TG synthesis.
28.4 Dyslipidemic Therapies
28.4.1 HMG-CoA Reductase Inhibitors
The statin classes of drugs, which reduce cholesterol biosynthesis by inhibiting its rate-limiting enzyme, 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase, are by far the most widely prescribed hypolipidemic drugs. As cited above, numerous trials have supported plasma cholesterol lowering with statins. However, statins have another widely studied cardioprotective effect, modulation of endothelial function.
Endothelial nitric oxide synthase (eNOS) forms nitric oxide (NO), which when released into the vasculature has anti-inflammatory, antiplatelet, antiproliferative, and anti-migratory properties in vessels [79]. Endothelial dysfunction is characterized by impaired endothelium-dependent relaxation due to reduced vessel wall NO bioavailability with subsequent increased oxidative stress and reduced vasorelaxation–vasoconstriction balance [80]. Thus, eNOS is central to the regulation of cardiovascular function. There is growing evidence that statins have salutary endothelial effects via increased eNOS activity in the context of atherosclerosis. For brevity, we will refer to statins generically rather than as a specific molecule even though they may elicit different effects if compared. The main endothelial effects of statins are as follows:
Reversal of tumor necrosis factor-α-induced downregulation in eNOS activity by inhibiting HMG-CoA reductase and blocking isoprenoid synthesis [81]
Induction of eNOS expression of LDL-exposed endothelial cells and inhibited lipid peroxidation [82]
Prevention of induced vascular endothelial dysfunction in rats by increasing NO bioavailability [83]
Prevention of induced vascular endothelial dysfunction in rats by enhancing the vascular NO generation, reducing the oxidative stress, and improving endothelial integrity and function [84]
Prevention of ischemia–reperfusion-induced myocardial injury by upregulating myocardial eNOS expression [85]
In addition, rosuvastatin increased vascular endothelial nitric oxide production and subsequently attenuated myocardial necrosis following ischemia and reperfusion in the mouse [85]. Genetic ablation of the eNOS gene abrogates the cardioprotective effects of a statin, thus implicating statin effects on eNOS in the protective effect [86].
28.4.2 Fibrates
Fibrates reduce plasma TG concentrations, which has the effect of raising plasma HDL-C concentrations [76]. The primary effect of fibrates is likely to increase hepatic β-oxidation; this reduces the amount of fatty acids available for TG synthesis, which lowers the hepatic secretion rate and the plasma concentration. Fibrates activate PPAR (peroxisome proliferator-activated receptors), mainly PPARα, a nuclear receptor that modulates carbohydrate and fat metabolism. Following activation by fibrates and some other fatty acids, PPARα heterodimerizes with the retinoid X receptor (RXR) bound to the PPAR-response element and activates the expression of downstream genes that code for proteins involved in mitochondrial and peroxisomal β-oxidation. The mechanism, shown in Fig. 28.3, is as follows: Fibrates bind to a PPARα and RXR heterodimer that is associated with the PPAR-response element within the promotor on a DNA strand thereby activating transcription of downstream genes, including those that promote enhanced β-oxidation. Increased β-oxidation reduces intracellular fatty acid concentrations and in hepatocytes reduces production and secretion of VLDL-TG.
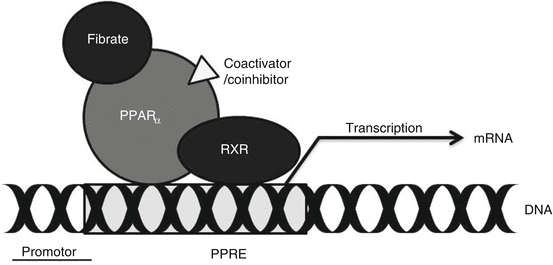
Fig. 28.3
Mechanistic model for the activity of fibrates. Fibrates bind to a PPARα and RXR heterodimer; the heterodimer binds to the PPAR-response element (PPRE) within the promotor for a specific gene thereby activating transcription of downstream genes
28.4.3 Cholesterol Absorption Inhibition
Ezetimibe lowers plasma cholesterol levels by inhibiting intestinal cholesterol absorption. Ezetimibe localizes to the small intestine brush border, where it binds to the Niemann–Pick C1-like 1, which mediates cholesterol absorption by gastrointestinal tract epithelial cells [13]. The reduced cholesterol absorption upregulates LDL receptors, mainly on hepatocytes, which increases LDL-C uptake thereby reducing plasma LDL levels. Although ezetimibe reduces LDL-C, it was not known to improve outcomes so that until ezetimibe is shown to have a clinical impact, it should be a second- or third-line agent [87].
28.4.4 Niacin
Niacin elicits its therapeutic effect by binding G protein-coupled receptors, niacin receptor 1 and 2, which are highly expressed in adipose tissue, but not in liver or intestine [88, 89]. Niacin receptor 1 inhibits cyclic adenosine monophosphate production and thus lipolysis in adipose tissue so that free fatty acids available for hepatic VLDL-TG production are reduced. As a consequence, the plasma product of VLDL hydrolysis, LDL-C, is also reduced. Reduced free fatty acids also suppress hepatic apo CIII expression and PPARγ coactivator-1b, thus increasing VLDL turnover and reducing its production [90]. The HDL-C-raising qualities may be due to reduced plasma concentration of TG no longer available for exchange for HDL-C. Notably, interest in niacin as a therapy was reduced by the AIM-HIGH Study, which was discontinued because of futility. Among atherosclerotic CVD patients with LDL-C levels <70 mg/dL, there was no incremental clinical benefit from the addition of niacin to statin therapy, despite improvements in HDL cholesterol and triglyceride levels [91]. The flushing inhibitor, laropiprant, is sometimes recommended to improve niacin tolerance.
28.4.5 Bile Acid Sequestrants
Bile acid sequestrants (BAS, resins) include cholestyramine, colestipol, and colesevelam. The former, cholestyramine, was used to demonstrate that reduction of serum cholesterol reduces CVD events in the National Institutes of Health Lipid Research Clinics Coronary Primary Prevention Trial [3, 92]. Its success provided a compelling rationale for the pursuit of more convenient and powerful cholesterol-lowering agents such as the statins. The sequestrants bind to bile salts in the intestine and prevent them from recycling to cholesterol, which would ultimately appear in plasma. Lowering hepatic bile acid levels stimulates the diversion of cholesterol to bile acid synthesis, which in turn increases hepatic cell-surface LDL receptor expression thereby providing an additional plasma LDL-C-lowering effect.
28.4.5.1 n-3 Fatty Acids
Fish oils, which are long-chain polyunsaturated n-3 fatty acids, are hypotriglyceridemic agents that also increase blood clotting time and improve vascular function. While reducing plasma TG levels, fish oils also raise HDL-C by a mechanism that involves reduced exchange of plasma HDL-C for VLDL-TG [76]. Fish oils, mostly as eicosapentaenoic and docosahexaenoic acids, can have an indirect therapeutic effect via their conversion to resolvins, via the COX-2 pathway especially in the presence of aspirin. Resolvins appear to reduce cellular inflammation by inhibiting the production and transportation of inflammatory cells and chemicals to the sites of inflammation [93]. Protectins, which have anti-inflammatory, anti-apoptotic, and neuroprotective properties, are formed by the hydroxylation of polyunsaturated fatty acids. For example, oxygenation of docosahexaenoic acid produces protectin D1, which has been attributed with multiple salutary effects [94–97], which may provide vascular protection. Other studies showed that dietary fish oil prevents vascular dysfunction in rats, an effect that is associated with increased eNOS expression and reduced oxidative stress [98].
28.4.5.2 CETP Inhibition
Much of the reduction in plasma HDL-C concentrations occurs via the exchange of HDL-C for VLDL-TG, which is mediated by cholesterol ester transfer protein (CETP). Moreover, populations with a defect in CETP activity have very high plasma HDL-C levels [99]. Thus, the negative correlation of plasma HDL-C levels and atherosclerotic CVD provided the impetus to inhibit this CETP activity pharmacologically. Several inhibitors were developed including torcetrapib and anacetrapib. Although the inhibitors increased plasma HDL-C levels [100], they were fraught with negative side effects (torcetrapib) [42] and/or did not improve CVD outcomes (dalcetrapib) [43]. Given that the occurrence of natural human CETP deficiency was not cardioprotective [101], the outcome did not surprise all.
28.4.5.3 Combination Therapy
Sometimes monotherapy with a statin is sufficient for reducing plasma LDL-C concentrations. However, combination therapy is an option for some patients in cases where high-statin doses are not tolerated and lower doses are not sufficient to bring LDL-C to target. In these instances, a combination of a statin and one of the other approved drugs—ezetimibe, fish oils, fibrates, or niacin—could be tried, with monitoring for efficacy.
The effects of lipid-lowering drugs on major plasma lipid analytes are given in Table 28.1, which also indicate possible side effects. Notably, the lipid-modifying effect is expected to be a function of dosing, formulation, and the specific drug within a class.
Table 28.1
Effects of major lipid therapies on plasma lipoprotein concentrations
Drug/class | Plasma lipid analytea | Adverse effects | ||
|---|---|---|---|---|
HDL-C | LDL-C | TG | ||
Statins | + | ─ ─ ─ | ─ | Myopathy, rhabdomyolysis, elevated liver enzymes, diabetes |
Fibrates | + | ─ | ─ ─ ─ | Myopathy, rhabdomyolysis |
n-3 fatty acids | ─ ─ | Increased blood clotting time, fishy taste | ||
Niacin | + | ─ | ─ ─ ─ | Flushing |
Ezetimibe | ─ | |||
Bile acid sequestrants | + | ─ | Constipation, fecal impaction | |
CETP inhibitors | +++ | CVD outcomes unproven | ||
Nutrition Therapy
Nearly all recommendations for the control of plasma lipid concentrations include a dietary component. Recommendations from the American College of Cardiology/American Heart Association (ACC/AHA) [102] and the American Diabetes Association [103] have shifted away from standard meal plans to eating patterns that are individualized and based upon each patient’s profile. This includes their health goals in the context of their personal and cultural preferences, health literacy, access to health-promoting foods, and the ability and commitment to changing eating patterns. There is an increased emphasis on appropriate portion size and reduced intake of processed nutrient-dense foods. In addition, ACC/AHA classifies the dietary recommendations according to the estimated certainty of a treatment effect that is weighed against a patient’s risk–benefit balance. These classifications extend from treatments with compelling evidence of a treatment effect and a high risk–benefit ratio that would warrant a recommendation to the other extreme, treatments with little evidence of support and a minimal risk–benefit ratio for which the treatment may be considered but its effectiveness is not well established. In some cases, where there is at least moderate evidence-based certainty that there is no net benefit or that risks/harm outweigh benefit, the treatment would not be recommended.
28.5 Concluding Remarks
According to the studies of various interventions superimposed on statin therapy, there remains a need to reduce residual CVD risk. It may be time to think outside the proverbial box and explore other possibilities. The area of research that is most compelling for further investigation is the relationship between energy metabolism and atherogenesis. This includes more research into the metabolic pathways and mechanisms that underlie the cardioprotective effects of exercise and of moderate alcohol consumption, both of which raise HDL-C and reduce CVD risk. Identification of their attendant pathways and mechanisms is important because the CVD risk reduction may occur via mechanisms that are parallel to but do not involve pathways that increase HDL-C.
References
1.
Kochanek KD, Xu J, Murphy SL, Minino AM, Kung HC. Deaths: final data for 2009. Natl Vital Stat Rep Centers Dis Control Prev Natl Center Health Stat Natl Vital Stat Syst. 2011;60:1–116.
2.
3.
The lipid research clinics coronary primary prevention trial results. I. Reduction in incidence of coronary heart disease. JAMA. 1984;251:351–64.
4.
Endo A. The discovery and development of HMG-CoA reductase inhibitors. J Lipid Res. 1992;33:1569–82.PubMed
5.
Brown AG, Smale TC, King TJ, Hasenkamp R, Thompson RH. Crystal and molecular structure of compactin, a new antifungal metabolite from penicillium brevicompactum. J Chem Soc Perkin 1. 1976;(11):1165–70.
6.
Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA, Langendorfer A, Stein EA, Kruyer W, Gotto Jr AM. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TEXCAPS. Air force/Texas coronary atherosclerosis prevention study. JAMA. 1998;279:1615–22.CrossRefPubMed
7.
Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the scandinavian simvastatin survival study (4s). Lancet. 1994;344:1383–9.
8.
Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, Brown L, Warnica JW, Arnold JM, Wun CC, Davis BR, Braunwald E. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events trial investigators. N Engl J Med. 1996;335:1001–9.CrossRefPubMed
9.
10.
Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland coronary prevention study group. N Engl J Med. 1995;333:1301–7.CrossRefPubMed
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


