Fig. 6.1
Signal transduction pathways and kinases involved in phosphorylation of cardiac titin N2-Bus and PEVK. AC adenylate cyclase, ßAR beta-adrenergic receptor, AngII angiotensin II, ANP atrial natriuretic peptide, ATP adenosine triphosphate, cAMP cyclic adenosine monophosphate, BNP brain natriuretic peptide, CaMKIIδ Ca2+/calmodulin-dependent protein kinase II delta, CNP natriuretic peptide, ET-1 endothelin-1, ERK2 extracellular signal-regulated kinase 2, G small G-protein, GTP guanosine triphosphate, cGMP cyclic guanosine monophosphate, GPCR G-protein coupled receptor, sGC soluble guanylyl cyclase, pGC peripheral guanylyl cyclase, IRb insulin receptor b, NO nitric oxide, PKA cAMP-dependent protein kinase, PKC Ca2+-dependent protein kinase, PKG cGMP-dependent protein kinase
The sequence insertions Zis-1 and Zis-5 at the NH2-terminal end of titin contain various consensus sequences for proline-directed kinases, the so-called XSPXR motif repeats. These motifs were shown to be phosphorylated in vitro by extracellular signal-regulated kinases 1 and 2 (ERK1/2) and cyclin-dependent protein kinase-2 (Cdc2) (Gautel et al. 1996; Sebestyén et al. 1995). Four KSP-phosphorylation motifs were identified in the sequence insertion Mis-4 in the COOH-terminal part of M-band titin. Similar to the XSPXR repeats in Z-disk titin, the KSP motifs are phosphorylated by proline-directed kinases (Gautel et al. 1993). Phosphorylation of these motifs has been demonstrated to occur mainly in developing rather than differentiated adult muscles and may therefore be required for a proper sarcomeric integration of titin during myofibrillogenesis (Sebestyén et al. 1995; Gautel et al. 1993).
The M-band region of titin contains an autoinhibited serine/threonine kinase domain, which shares some homology with Ca2+/calmodulin-regulated myosin light chain kinases (MLCK) (Gautel 2011). Activation of TK requires the removal of the C-terminal autoinhibitory tail; however, unlike other classical MLCKs the modulation of TK activity by Ca2+-Calmodulin is rather weak (Mayans et al. 1998). Whether activation of the TK instead involves autophosphorylation of sites in the activation loop of the kinase is still under debate. It has further been proposed from molecular dynamics simulations that activating changes to the conformational state of the TK could be induced by mechanical stretch forces (Gräter et al. 2005; Puchner et al. 2008). Upon activation TK has been shown to interact with the ubiquitin-associated zinc-finger protein neighbor-of-BRCA1-gene-1 (Nbr1), which forms a signaling complex with p62/SQSTM1 and the muscle-specific ubiquitin E3 ligases MuRF1, MuRF2, and MuRF3 (Lange et al. 2005). Due to this interaction, TK has been suggested to be an important biomechanical stress sensor and mediator of hypertrophic signaling (Gautel 2011).
6.2.2 I-Band Phosphorylation of Titin
The main hub for titin phosphorylation is the elastic I-band region, particularly the N2-B region with its unique sequence (us) and the PEVK domain. Phosphorylation of these two titin domains dynamically modulates the passive mechanical properties of the myofilaments (Fig. 6.1).
CAMP-dependent protein kinase (PKA) has been shown to phosphorylate the cardiac-specific N2-B domain and thereby reduce passive tension in rat, bovine, and human cardiomyocytes (Yamasaki et al. 2002; Fukuda et al. 2005; Krüger and Linke 2006) (Fig. 6.2a). In human myocardium, PKA and cGMP-dependent protein kinase (PKG) both phosphorylate a serine residue in the cardiac-specific N2-Bus (Ser4185, UniProtKB accession number, Q8WZ42). Single molecule experiments using a recombinant construct of this region demonstrated that phosphorylation increases the persistence length of the N2-Bus and thereby reduces titin-based passive tension (PT) by up to 20 % (Krüger et al. 2009) (Fig. 6.2a). More recent studies applying mass-spectrometry analyses on kinase-treated recombinant titin fragments have identified further sites of PKA- and PKG-mediated phosphorylation in the N2-Bus, some of which are conserved among species (Kötter et al. 2013; Hamdani et al. 2013c) (an overview on currently identified I-band phosphorylation sites is provided in Table 6.1). The presence of multiple kinase-dependent phosphorylation motifs within this region suggests a mechanism by which titin stiffness is rapidly modulated via simultaneous hypo- or hyperphosphorylation of these sites, e.g., in response to beta-adrenergic stimulation.
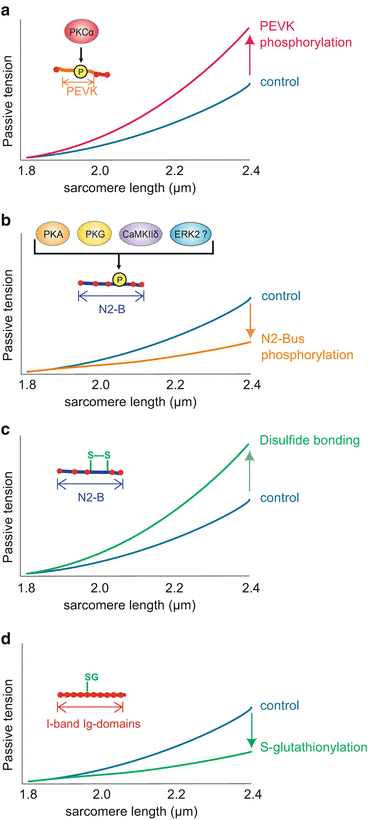
Fig. 6.2
Posttranslational modification of titin and effects on myofilament stiffness. (a) Phosphorylation of the PEVK region of titin by PKCα increases titin-based myofilament stiffness; (b) phosphorylation of the N2-B region of titin by PKA, PKG, ERK and CaMKIIδ decreased myofilament stiffness; (c) disulfide bonding within the N2-B region of titin increases titin-based myofilament stiffness; (d) S-glutathionylation (SG) of unfolded Ig domains lowers titin stiffness
Table 6.1
I-band phosphosites identified in titin N2-Bus/PEVK
Phosphosite | Titin region | Species | Kinase | Reference |
|---|---|---|---|---|
Ser3744 | N2-Bus | Rat | PKA/PKG | Kötter et al. (2013) |
Ser3750 | N2-Bus | Conserved | CaMKIIδ | Hidalgo et al. (2013) |
Thr3756 | N2-Bus | Human | CaMKIIδ | Hamdani et al. (2013c) |
Thr3761 | N2-Bus | Human | CaMKIIδ | Hamdani et al. (2013c) |
Ser3787 | N2-Bus | Human | CaMKIIδ | Hamdani et al. (2013c) |
Ser3799 | N2-Bus | Human | CaMKIIδ | Hamdani et al. (2013c) |
Ser3862 | N2-Bus | Human | CaMKIIδ | Hamdani et al. (2013c) |
Thr3864 | N2-Bus | Human | CaMKIIδ | Hamdani et al. (2013c) |
Ser3918 | N2-Bus | Human | ERK2 | Raskin et al. (2012) |
Ser3960 | N2-Bus | Human | ERK2 | Raskin et al. (2012) |
Ser4010 | N2-Bus | Conserved | PKA | Kötter et al. (2013) |
ERK2 | Raskin et al. (2012) | |||
Ser4012 | N2-Bus | Conserved | PKA | Kötter et al. (2013) |
Ser4062 | N2-Bus | Conserved | CaMKIIδ | Hamdani et al. (2013c) |
Ser4065 | N2-Bus | Human/dog | PKA | Kötter et al. (2013) |
Thr4065 | N2-Bus | Rat/mouse | PKA | Kötter et al. (2013) |
Ser4092 | N2-Bus | Human/dog | PKG | Kötter et al. (2013) |
Ser4099 | N2-Bus | Conserved | PKG | Kötter et al. (2013) |
CaMKIIδ | Hamdani et al. (2013c) | |||
Ser4185 | N2-Bus | Human | PKA/PKG | |
Thr4203 | N2-Bus | Human | CaMKIIδ | Hamdani et al. (2013c) |
Ser4209 | N2-Bus | Conserved | CaMKIIδ | Hidalgo et al. (2013) |
Ser11878 | PEVK | Conserved | PKCα | |
CaMKIIδ | Hidalgo et al. (2013) | |||
Thr11922 | PEVK | Human | CaMKIIδ | Hamdani et al. (2013c) |
Thr11932 | PEVK | Human | CaMKIIδ | Hamdani et al. 2013c |
Thr11969 | PEVK | Human | CaMKIIδ | Hamdani et al. (2013c) |
Thr12007 | PEVK | Conserved | CaMKIIδ | Hamdani et al. (2013c) |
Ser12009 | PEVK | Conserved | CaMKIIδ | Hamdani et al. (2013c) |
Ser12022 | PEVK | Conserved | PKCα | |
CaMKIIδ | Hamdani et al. (2013c) |
In vitro kinase assays using recombinant fragments of different I-band domains of titin have shown PKG-mediated phosphorylation of several Ig domains and the N2-A domain of titin (Krüger et al. 2009). Since modification of these domains does not contribute to titin-based PT under physiological conditions (Krüger et al. 2009), the functional role of these phosphorylation motifs remains to be elucidated. The N2-A region associates with muscle ankyrin repeat proteins (MARPs) and in skeletal muscle with Calpain-3, thereby linking titin to hypertrophic signaling and the protein quality control machinery (Miller et al. 2003; Hayashi et al. 2008). Hence, phosphorylation of the N2-A region could alter protein–protein interactions and thereby modulate the respective signaling pathways.
PKA shares a conserved phosphorylation site (Ser4010) with ERK2, which has been shown to target the N2-Bus of titin at three different serine residues (see Table 6.1) (Raskin et al. 2012). Although it has not been confirmed experimentally, yet, this ERK2-mediated phosphorylation is expected to result in a reduction in titin-based PT. Interestingly, titin also associates with the four-and-a-half LIM domain proteins FHL-1 and FHL-2 in the N2-B region (Sheikh et al. 2008; Lange et al. 2002) and is thereby linked to the Ras-Raf-MEK-ERK pathway, which has an important cardioprotective role (reviewed in Wang 2007). In the absence of biomechanical stress, MEK1/2 act as scaffold proteins for ERK2 and anchor the kinase in the cytoplasm (Tanoue et al. 2000; Chuderland and Seger 2005). However, it may be speculated that the titin/FHL/MEK complex tethers a certain portion of ERK2 to the myofilaments. In response to mechanical strain, MEK then activates ERK2, which induces a conformational change that promotes its translocation to the nucleus, where ERK2 is responsible for activation of several transcription factors involved in hypertrophic growth (Mebratu and Tesfaigzi 2009). Interestingly, disruption of the FHL-Raf-MEK-ERK-signaling complex by genetic knockdown of FHL1 in mice resulted in increased myofibrillar compliance and impaired hypertrophic signaling (Sheikh et al. 2008). ERK2-mediated phosphorylation may therefore be another regulatory mechanism that contributes to the biomechanical stress sensing mediated at the N2-Bus of titin.
Finally, phosphorylation of the N2-B region is induced by Ca2+/calmodulin-dependent protein kinase II delta (CaMKIIδ). Using mass spectrometry ten different serine and threonine residues have been identified as putative CaMKIIδ targets in the N2-Bus (Hidalgo et al. 2013; Hamdani et al. 2013c). CaMKIIδ-mediated phosphorylation of single murine cardiomyocytes markedly reduced titin-based passive tension by up to 50 % (Fig. 6.2a). This finding is in line with the observation that phosphorylation of the CaMKIIδ-targeted phosphosites and titin-based PT is significantly reduced in CaMKIIδ-depleted mice (Hamdani et al. 2013c).
Interestingly, mass spectrometry further identified seven CaMKIIδ-targeted phosphorylation motifs in the PEVK region (Hidalgo et al. 2013; Hamdani et al. 2013c), another elastic titin domain that is functionally modified by phosphorylation (Table 6.1). Previous studies had shown that phosphorylation of Ser11878 and Ser12022 in the PEVK region by Ca2+-dependent protein kinase C α (PKCα) reduces the persistence length of the respective PEVK peptide and thereby increases titin stiffness by >20 % in skinned myocytes from mouse and pig heart (Hidalgo et al. 2009; Hudson et al. 2010) (Fig. 6.2b). Interestingly, although the identified PKCα-targeted serine residues are conserved among these species, this effect was not observed in cardiomyocytes from dog heart (Hamdani et al. 2013a).
The observation that phosphorylation of the N2-Bus by PKA, PKG, or CaMKIIδ leads to an increase of the persistence length and decreases myofilament passive tension, whereas phosphorylation of the PEVK domain by PKCα reduces the persistence length of the respective region and increases passive tension, may seem rather contradictory at first sight. A possible explanation is based on the amino acid sequences of the respective titin segments: The N2-Bus contains a high amount of acidic (negatively charged) amino acids, resulting in a low isoelectric point of this domain. In contrast, the PKCα-targeted region of the PEVK domain comprises many basic residues and therefore has a much higher isoelectric point than the N2-Bus. Introduction of a negatively charged phosphate group into an already negatively charged environment could lead to intramolecular electrostatic repulsion. This, in turn, could influence the intrinsically disordered structure of the N2-Bus and thereby increase its distensibility, reflected by an increased persistence length. In the PEVK region, the negatively charged phosphate residues are added to a basic environment, which could prevent electrostatic repulsion and potentially generate additional intramolecular ionic interactions. Such interactions likely reduce the persistence length of the PEVK domain and therefore impair its distensibility. According to this view, phosphorylation of several residues within each region should multiply the phosphorylation-induced effect on the molecular elasticity of titin and thereby allow a dynamic and presumably nonlinear modulation of titin stiffness (Kötter et al. 2013).
This hypothesis also implies that the effect of a few phosphorylated residues in one titin domain, e.g., in the PEVK region, may be counterbalanced or even offset by hypophosphorylation of the other elastic titin domain, e.g., the N2-Bus. This consideration may also provide an explanation for the observation that CaMKIIδ-mediated phosphorylation of titin results in a decrease in titin stiffness, although the kinase targets both elastic domains, N2-Bus and PEVK (Hamdani et al. 2013c).
Analysis of titin phosphorylation is an emerging field and online routines (e.g., GPS, Scansite, NetphosK) predict phosphorylation of the titin sequence at numerous additional sites and by various other kinases. Therefore, identification of more phosphorylation motifs and characterization of their functional relevance seem just a matter of time.
6.3 Regulation of Titin Phosphorylation in Health and Disease
6.3.1 Altered Titin Phosphorylation in Heart Failure
The cellular mechanisms that regulate titin function via domain-specific phosphorylation are only beginning to be understood. However, the dynamic modulation of titin does not only provide possibilities for its adaptation to various physiological conditions, but also represents a potential target for disturbances in the setting of cardiac diseases. It is therefore not surprising that there is a growing body of evidence showing altered titin phosphorylation in different types of heart failure.
First evidence for PKG-dependent hypophosphorylation of total titin in failing hearts was provided from PKG back-phosphorylation assays performed on heart samples from patients with dilated cardiomyopathy (DCM) (Krüger et al. 2009). This finding was supported by a study that, by using the phosphospecific protein stain ProQ-Diamond (Invitrogen), demonstrated a hypophosphorylation of total titin and raised cardiomyocyte resting tension in biopsies of heart failure (HF) patients (Borbely et al. 2009). Interestingly, PKA treatment of skinned cardiac myocytes from these samples effectively lowered passive myocyte stiffness to that of control cells (Borbely et al. 2009).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


