Name of enzyme
Major cardiac phenotypes
Related mechanisms
References
HDAC1 & 2
Double KO: Dilated cardiomyopathy, arrhythmias, and premature death
HDAC1 and 2 are functionally redundant. Double KO dysregulates the expression of genes encoding ion channels and skeletal muscle contractile proteins
[81]
HDAC3
KO: Variety of cardiac structural defects including double-outlet right ventricle, ventricular septal defects, interrupted aortic arch type B
Overexpression: Increased postnatal cardiomyocyte proliferation without hypertrophy
Defective smooth muscle differentiation, decreased expression of the notch ligand Jaged1
HDAC2
KO: Attenuated cardiac hypertrophy in response to hypertrophic stimuli
Transgenic: augmented hypertrophy
Modulating PI3K-Ak5-Gsk3b signaling pathway
[78]
HDAC4
KO: no obvious cardiac phenotypes
[84]
HDAC5 & 9
Single KO: exaggerating hypertrophic response to pressure overload but not to chronic b-adrenergic stimulation
Compound KO: embryonic/early postnatal death with ventricular septal defects and thin ventricular wall
HDAC5 and 9 are functionally redundant. HDAC9 mediates the transcriptional activity of MEF2
HDAC6
KO: attenuating the cardiac dysfunction in response to AngII stimulation.
Unclear
[85]
HDAC7
KO: embryonic lethality due to blood vessel dilation and rupture
Suppressing the expression of matrix metalloproteinase 10 via physical interaction with MEF2
[86]
HDAC8
KO: Global KO mice exhibited no discernible cardiac phenotypes but had skull defects
Dysregulation of homeobox genes in cranial neural crest cells
[87]
HDAC11
KO: No discernible gross phenotypes; had some effects on immune system
Unclear
[88]
14.2.5 HDAC Inhibitors in Cardiac Hypertrophy and Heart Failure
The development and application of HDACs inhibitors in the treatment of human cardiomyopathy and heart failure may be one of many therapeutic strategies. Indeed, TSA, a chemical HDAC inhibitor, suppressed myocardial fibrosis in murine models of cardiac hypertrophy induced by either aortic banding or Hopx gene overexpression [91, 92]. Sodium valproate, another chemical HDAC inhibitor, ameliorated right ventricular hypertrophy induced by pulmonary artery banding in a rat model [93]. Some excellent reviews are recommended for those interested [94, 95]. In all, these findings may hold promise for the clinical application of HDAC inhibitors in cardiac diseases.
14.3 Ubiquitination and Cardiac Function/Diseases
Ubiquitin, a highly conserved 76 amino acid protein (~16kd), which was discovered in the 1970s [96, 97], is the founding member of the super family of ubiquitin-like proteins or modifiers. Ubiquitin can be covalently conjugated to its targets via a series of chemical cascades that requires activating enzyme E1, conjugating enzyme E2 and ligases E3 (Fig. 14.1), consequently either causing targets’ degradation or altering targets’ activity, depending on the position of lysine that is used for covalent ubiquitin-ubiquitin linkage [98]. For instance, the substrates with polyubiquitination formed on lysine 48 of ubiquitin are usually delivered to proteasome complex for proteolysis; however, monoubiquitination or polyubiquitination linked on lysine 63 of ubiquitin usually alters the activity of substrates (Fig. 14.1). Ubiquitination is a reversible process that can be de-ubiquitinated by a group of enzymes [99]. Selectivity of ubiquitination substrates is mainly governed by several hundred E3 ligases that have been identified up to date; however, only a few are muscle-specific E3 ligases and will be discussed in this chapter regarding their association in heart diseases.
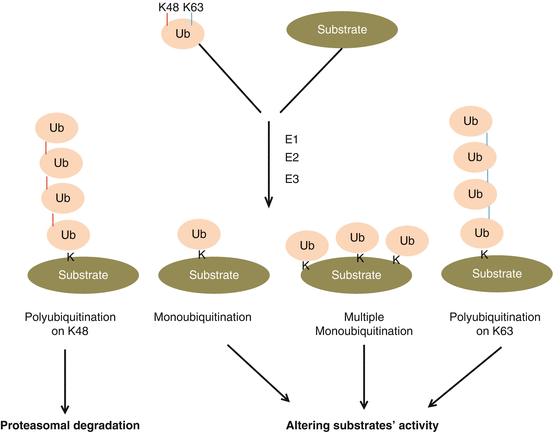
Fig. 14.1
The conjugation of ubiquitin. Ubiquitination involves activating enzyme E1, conjugating enzyme E2 and ligase E3. Up to date single E1, a few E2, and couple hundred E3 have been identified, and E3 mainly determines the substrate specificity of ubiquitination. Ubiquitination may have several different forms: polyubiquitination on lysine 48, mainly resulting in proteasomal degradation of substrates, and polyubiquitination on lysine 63 of ubiquitin or monoubiquitination (one ubiquitin attached to one lysine on the substrate), or multiple monoubiquitination (multiple ubiquitins attached to lysines on different locations of substrate protein), consequently resulting in the change in the function of substrates
14.3.1 Ubiquitin Proteasome System and Heart Diseases
The proteasome is composed of a large multienzymatic protease complex that degrades the vast majority of intracellular proteins, including nonfunctional (misfolded or damaged) or functional proteins in an ATP-dependent manner. The major components of ubiquitin proteasome system (UPS) are the 26S proteasome and ubiquitin. Polyubiquitinated proteins formed on lysine 48 on the ubiquitin molecule are usually targeted for proteasome-mediated degradation. Recent studies indicate an important role for UPS in cardiac diseases.
Familial Dilated Cardiomyopathy
The familial dilated cardiomyopathy associated with UPS is the desmin-related myopathy. Desmin is a filament protein essential for normal cardiomyocyte structure, and its stability is mediated by a chaperon protein named αB-crystallin, a heat shock protein that recruits desmin for proteasome-associated degradation. Mutations of desmin and/or αB-crystallin, like p.Arg120Gly, substantially impair the function of αB-crystallin [100] and cause dilated cardiomyopathy and heart failure [101, 102]. This human dilated cardiomyopathy was recapitulated in the mouse model in which αB-crystallin-Arg120Gly mutant was specifically expressed in cardiomyocytes. The transgenic mice developed mitochondrial disorganization and dysfunction and cardiomyopathy and eventually were succumbed to heart failure [103]. Further studies revealed the impairment of UPS and increased aberrant protein aggregates underlying the development of cardiomyopathy [104], indicative of the implication of UPS in desmin-related cardiomyopathy.
Familial Hypertrophic Cardiomyopathy
The most common familial hypertrophic cardiomyopathy is caused by the mutations on MYBP3 gene, which encodes cardiac myosin-binding protein C (cMBP-C) [105] (see Chap. 60). Most mutations on MYBP3 generate truncated proteins [106], likely degraded by UPS [107], thus increasing the UPS workload. Indeed, impaired UPS was observed in the cultured cardiomyocytes with adenoviral-mediated expression of truncated cMBP-C [107], and transgenic mice overexpressing truncated cMBP-C in heart developed cardiac hypertrophy as expected [108].
Ischemic Heart Disease
The suppression of UPS activity by coronary ischemia/reperfusion was first reported in 2001 [109] and later confirmed [110]. Decreased UPS activity led to increased accumulation of abnormal protein aggregates, which contributes to cardiac dysfunction after ischemic injury. Correspondingly, overexpression reduced the aggregation of aberrant proteins, infarct size, and improved cardiac function after ischemia/perfusion [111]. In contrast, inhibition of the proteasome activity in cardiomyocytes heightened the myocardial ischemia/reperfusion injury in mice [112]. Mechanistically, increased reactive oxidative species and decreased ATP content may underlie dysfunctional UPS activity observed in the ischemia/reperfusion heart [110, 113].
Cardiac Remodeling and Heart Failure
Cardiac remodeling occurs in response to elevated workload such as hypertension or pathological stimuli, such as ischemia, and may progress to heart failure, characteristic of reduced cardiac pump function. Although the implication of UPS in cardiac remodeling and heart failure has been previously studied, the findings are confounding. The UPS activity was reported to be deficient in some studies but to be elevated in others on the human failing hearts and/or in the animal models with cardiac remodeling and heart failure [114–116]. Recently, a selective instead of a general suppression of the proteasomal function caused by oxidative injury [117] further complicates the issue. More extensive studies will be needed to clarify these issues.
14.3.2 Ubiquitination E3 Ligases and Cardiac Diseases
As mentioned above, the specificity and selectivity of substrates for UPS-mediated degradation are governed by ubiquitination E3 ligases, which are divided into three classes: the RING finger-containing proteins, the U-box-containing proteins, and the HECT-domain-containing proteins. The major cardiac ubiquitin E3 ligases that have been shown to play a role in cardiac physiology and pathophysiology include atrogin-1, the muscle RING finger (MuRF) family (MuRF1, 2, and 3), carboxyl-terminus of heart shock protein 70-interacting protein (CHIP), and murine double minute 2 (MDM2).
Atrogin-1
Atrogin-1 (atrophy gene 1, or muscle atrophy F-box, MAFbx) harbors an F-box motif, characteristic of functional domain identified in the components of the SCF (skp1, cullin, F-box protein) ubiquitin ligase complex. Atrogin-1 is specifically expressed in the cardiac and skeletal muscles in humans, mice, and rats [118, 119], thus defined as a muscle and cardiac-specific E3 ligase. Atrogin-1 interacts with the SCF ubiquitin ligase complex and brings its substrates for degradation. One of atrogin-1’s substrates is calcineurin. Calcineurin plays an important role in cardiac pathological hypertrophy in response to pressure overload and pro-hypertrophic stimuli [120]. Overexpression of atrogin-1 promoted degradation of calcineurin and repressed NFATc4 nuclear translocation, inhibiting cardiac hypertrophy induced by pressure overload [121]. However, another study showed that expression of atrogin-1 was substantially elevated in heart by pressure overload and that knockout of atrogin-1 was found to repress the cardiac hypertrophy induced by both pressure overload and β-adrenergic activation [122]. Mechanistically, stabilization of IkB-a and repression of NF-kB underlies this protective effect rendered by atrogin-1 ablation [122]. Currently, additional work will be needed to reconcile the abovementioned conflicting studies.
MuRF1
MuRF1, a RING domain-containing E3 ligase, was found to play a critical role in mediating cardiac troponin 1 stability [123, 124]. An earlier study showed that MuRF1 null mice had hearts with comparable mass relative to wild-type mice measured at baseline but were more resistant to hypertrophy regression [125]. However, MuRF1 knockout mice developed physiological cardiac hypertrophy, and MuRF1 was not required for cardiac atrophy [126]. These discrepancies could be attributable to different experimental settings used in these two studies, such as age and sex of animals used, the duration of stimulation, etc. Also, overexpression of MuRF1 facilitated heart failure in response to aortic banding [127]. More importantly, MuRF1 expression levels were increased in the cardiac muscles obtained from the patients with therapeutic cardiac atrophy after left ventricular assist device placement [125], indicative of a potential role for MuRF1 in human heart diseases.
MDM2
MDM2 also harbors a RING domain like MuRF1. MDM2 expression levels were elevated in murine hearts challenged with oxidative stress and pressure overload [128]. Overexpression of MDM2 in cultured cardiomyocytes protected them from hypoxia-reoxygenation-induced injury, while suppression of MDM2 activity blocked its protective activity [129]. Several major ubiquitination targets whose degradation is mediated by MDM2 are p53 [130, 131], ARC (apoptosis repressor with caspase recruitment domain) [128], β2-adrenergic receptor and β-arrestin [132], insulin-like growth factor 1 receptor [133], and FoxO1 [134, 135].
CHIP
CHIP is a U-box-containing E3 ligase, and also a chaperone of the heat shock proteins (HSP) 70 and 90. CHIP is highly enriched in heart [136], and CHIP−/− mice showed high mortality and severe cardiac hypertrophy compared with wild-type mice [137]. Overexpression of CHIP in murine hearts was protected from ischemia-induced cell death [138], thus demonstrating the protective role of CHIP in the maintenance of cardiac homeostasis. As an E3 ligase, CHIP has some common targets for proteasome-induced degradation as MDM2, including p53 and FoxO1 [139, 140].
14.3.3 Deubiquitinase and Heart Disease
Deubiquitinases play an important role in balancing ubiquitination-deubiquitination and thus maintains stable protein homeostasis. Currently about 79 deubiquitinases have been predicted in the human genome [141]. The involvement of deubiquitinases in immune system and cancer has been previously studied [142, 143], in which suppressing deubiquitinase activity may be a potential therapeutics for cancer [144]. More recently, mutations on USP8, a deubiquitinase gene, were causally linked to human Cushing’s disease [145]. However, the information regarding the implication of deubiquitinases in heart diseases is quite limited. Analysis from heart samples obtained from human dilated cardiomyopathy indicated dysregulated expression of deubiquitination enzyme isopeptidase-T and the ubiquitin-fusion degradation system-1 [146], two important deubiquitinases. However, how these changes really contribute to the pathogenesis of cardiomyopathy remains obscure.
14.4 SUMO Conjugation and Cardiac Function/Diseases
SUMO (small ubiquitin-like modifier) belongs to the ubiquitin-like superfamily. So far four SUMO family members, named SUMO-1, SUMO-2, SUMO-3, and SUMO-4, have been identified; however, SUMO-4 appears not to be conjugatable in its natural form [147]. SUMO-1 shares ~50 % homology with SUMO-2 and SUMO-3, but the active SUMO-2 and SUMO-3 proteins exhibit ~97 % identify. SUMO-1 and SUMO-2/3 may have distinct roles in regulating cellular activities, because (a) they exhibit substrate specificity to some degree [148]; (b) SUMO-2/3 appears to be more responsive than SUMO-1 to external stimuli, at least in cultured cell lines [149]; and (c) SUMO-1 exhibits different subcellular localizations and dynamics than SUMO-2/3 [148].
Like ubiquitination, sumoylation is also an enzymatic process that requires E1 (heterodimeric), E2 (Ubc9), and E3 and is ATP dependent (Fig. 14.1). Sumoylation is also a reversible process. Sentrin-specific proteases (SENPs) are responsible for deconjugating SUMO conjugation and also responsible for maturation of initially synthesized inactive SUMO precursors [150]. SUMO E3 ligase may enhance the specificity and efficiency of SUMO conjugation (see below). It is noteworthy that sumoylation may occur on a single or multiple lysine residues of a given substrate, named mono- or multisumoylation, respectively (Fig. 14.2). SUMO moieties can also be conjugated to the lysine residue within a SUMO protein that is attached to a substrate, forming a polymeric (poly-)SUMO chain (i.e., polysumoylation). Given the transient and dynamic nature of SUMO conjugation and the fact that in most cases only a small fraction of a given substrate is modified in any given time frame, it has been a challenge to detect endogenous sumoylated targets. Although the majority of SUMO substrates identified so far is nuclear, a number of SUMO targets are also localized in other subcellular compartments such as cytoplasm, plasma membrane, and mitochondria [151–155].
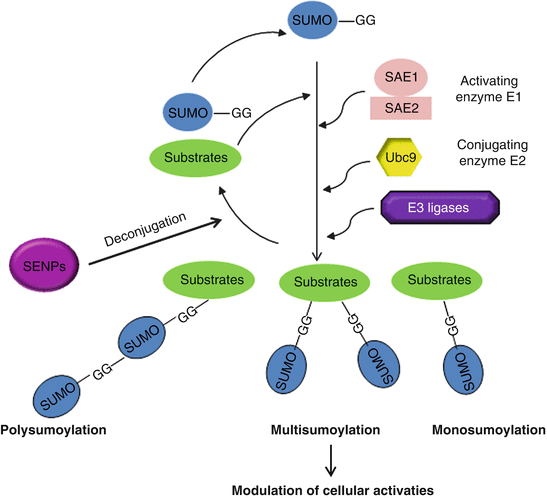
Fig. 14.2
SUMO conjugation machinery. Sumoylation requires heterodimeric activating enzyme (E1), conjugating enzyme (E2), and E3 ligase. SENPs deconjugate SUMO substrates. SUMO conjugation may occur in three forms: mono-, multi-, and polysumoylation
SUMO E3 Ligases
SUMO E3 ligases (Table 14.2) may catalyze SUMO conjugation by activating non-consensus SUMO acceptor site(s) and/or promoting multisumoylation/polysumoylation. SUMO E3 ligases are also believed to play a role in substrate discrimination. Among these known SUMO E3 ligases, PIAS family that consists of four isoforms (PIAS1, PIAS3, PIASx, and PIASy) is the most potent SUMO E3 ligase. However, the ligase activity of PIAS proteins might not always be attributable to the RING domain [166]. Of note, PIAS proteins themselves are SUMO substrates, although the SUMO acceptor sites remain to be identified [178, 179]. PIAS family members appear to have overlapping/redundant function, because knockout of PIAS1, PIAS2, or PIAS4 in mice did not lead to any severe and lethal phenotype(s) [180–183], while double knockout of PIAS1 and PIAS4 caused mouse embryonic lethality before E11.5 [184].
Desumoylation Enzyme SENPs
The family of SUMO-specific proteases (SENPs) consists of six members (SENP1, 2, 3, 5, 6, 7) in human that are specifically involved in the SUMO conjugation pathway [185]. SENPs cleave the C-terminal extensions of the SUMO proteins for maturation (endopeptidase activity) and free SUMO proteins that are attached to substrates (isopeptidase activity) (Fig. 14.1). SENPs differ in the capacity of their endopeptidase/isopeptidase activity as well as in their discrimination of substrates/SUMO isoforms [186, 187]. For instance, SENP1 and SENP2 exhibit both endopeptidase/isopeptidase activities for all SUMO precursors, while SENP3, 5, 6, and 7 preferentially target SUMO-2/3 [185]. Data for SENP3, 5, 6, or 7 knockouts are not available, but knockout of either SENP1 or SENP2 caused embryonic lethality [188–190], suggesting the non-redundant role of SENP proteins in murine embryonic development.
SUMO Targeting Sequence
SUMOs target the lysine (K) residue mainly localized in the known consensus sequence ψKXE/D (ψ is a bulky hydrophobic amino acid, X is any residue, D is aspartic acid, and E is glutamic acid) [191, 192]. However, sumoylation can also occur on noncanonical lysine residue(s) within some substrates [193, 194]. SUMO E3 ligases can induce the conjugation of SUMO to the noncanonical lysine residue(s) [195]. It is also worth noting that not all proteins that harbor this four-amino acid sequence motif are SUMO targeted. Interestingly, SUMO-2/3 also has a consensus sequence ValLysThrGlu that contains the lysine 11 [196], which is the critical site for poly-SUMO chain formation.
14.4.1 Involvement of SUMO Conjugation in Cardiovascular Function in Animal Models
The implication of SUMO conjugation in oncogenesis has been studied widely. Recent findings suggest that SUMO may be important for cardiac function and is involved in cardiovascular disorders. Indeed, SUMOs directly modify many substrates that are important for cardiovascular development and function (Table 14.3). For instance, Nkx2-5, Tbx5, and Zic3 are all important regulators for mouse cardiovascular development/function. While SUMO conjugation enhanced the activity of Tbx5 and Nkx2-5 [203, 219, 222], it repressed Zic3 function, at least partially, via modulating the nuclear localization of Zic3 [221], raising the possibility of a direct implication of SUMO conjugation in congenital heart diseases (CHDs). In support of this hypothesis, a subset of SUMO-1 null mice exhibited premature death with cardiac structural defects [223, 224], although this phenotype was likely affected by genetic background [225, 226]. The importance of SUMO-1 conjugation in early heart development was further highlighted by the observation that overexpression of SENP2, a pan-SUMO isopeptidase in mouse hearts, decreased SUMO conjugation and caused cardiac structural abnormalities as well as early demise, and simultaneously cardiac overexpression of SUMO-1 rescued heart defects and decreased the mortality rate of SENP2 transgenic mice [227].
Table 14.3
SUMO substrates important for cardiovascular development/function
SUMO targets | Major sumoylation site(s) | References |
|---|---|---|
Connexin 43 | Lysine 144/237 | [151] |
Drp1 | Multiple lysine residues | |
Erk5 | Lysine 6/22 | [197] |
ERRα | Lysine 14/403 | [198] |
Ezh2 | N/A | [199] |
GATA4 | Lysine 366 | [167] |
Lamin A | Lysine 201 | [200] |
Mef2c | Lysine 391 | [157] |
Msx1 | Lysine 9/127 | [201] |
Myocardin | Lysine 445 | [195] |
Nfatc1/C | Lysine 349/702/914 | [202] |
Nkx2-5 | Lysine 51 | [203] |
PARP1 | Lysine 203/486 | [204] |
PGC-1α | Lysine 183 | [205] |
PPARγ | Lysine 107 | |
PPARα | Lysine 385 (murine) Lysine 185 (human) | |
Prox1 | Lysine 556 | |
Retinoid x receptor α | Lysine 108 | [213] |
SERCA2a | Lysine 480/585 | [214] |
Smad4 | Lysine 113/159 | |
SRF | Lysine 147 | |
Tbx5 | N/A | [219] |
TRPM4∂ | Non-identified | [220] |
Zic3 | Lysine 248 | [221] |
In the SENP2 gain-of-function model, the conjugation of SUMO-1, SUMO-2, and SUMO-3 was diminished due to upregulated desumoylation activity [227]. Although a subset of SENP2-Tg mice died from CHDs, the surviving SENP2-Tg mice developed cardiac hypertrophy with age, accompanied by impaired cardiac function and elevated fibrosis [227], implicating the SUMO conjugation pathway in the development of cardiomyopathy. It will be interesting to dissect the role of SUMO-1 from that of SUMO-2/3 in development of cardiomyopathy, if any.
14.4.2 Potential Implication of SUMO Conjugation in Cardiovascular Diseases in Humans
14.4.2.1 SUMO and CHDs
Accumulating evidence points to a role of the SUMO conjugation pathway in cardiovascular disorders in humans. The abovementioned SUMO substrates such as Nkx2-5 and Zic3 all are linked etiologically to human congenital heart defects. A number of disease-linked naturally occurring Nkx2-5 and Zic3 mutants showed significantly decreased SUMO conjugation [221, 228]. Cardiac-specific expression of the Nkx2-5 SUMO site mutant (lysine 51 to arginine, Lys51Arg) in wild-type mice did not cause severe phenotype(s); however, it did induce CHDs in Nkx2-5 haploinsufficient mice (Nkx2-5+/−) [228]. Given the comparable expression level of the transgene K51R and wild-type Nkx2-5 in compound transgenic Lys51Arg:Nkx2-5+/− mouse hearts and the decreased sumoylation level of a number of human Nkx2-5 mutants, there is a likelihood that the altered sumoylation of Nkx2-5 contributes to the development of CHDs associated with these Nkx2-5 mutants. Another interesting finding is that sequencing analysis of the SUMO-1 gene of control and newborn patients with both cleft lip/palate and atrial septal defects (ASDs) reveals mutations in a cis-regulatory element of the SUMO-1 gene, mutations that repressed the reporter activity by ~50 % in the in vitro assays [224]. However, to further link SUMO-1 gene with human genetic diseases, more human samples will be needed for analysis, and family pedigree studies will also be required.
14.4.2.2 SUMO and Cardiomyopathy
Mutation of lamin A, a nuclear structural protein, is implicated etiologically in inherited dilated cardiomyopathy [229] (see Chap. 59). SUMO modifies the lysine residue 201 in a consensus sequence MetLysGluGlu of lamin A and the mutation of glutamic acid 203 to either glycine (Glu203Gly) [230] or lysine (Glu203Lys) [231] (which are associated casually with familial dilated cardiomyopathy and conduction disease), negatively affects lamin A sumoylation leading to altered nuclear localization [200]. These two mutations displayed similar molecular phenotypes exhibited by the sumoylation-deficient Glu201Arg mutation [200]. Thus, deficient sumoylation of lamin A is implicated directly in familial dilated cardiomyopathy.
SERCA2a, an ATPase and a critical factor in handling Ca2+ homeostasis during excitation-contraction coupling, was identified as a SUMO substrate on two sites: K480 and K585 [214]. The levels of both free and SUMO-1 conjugated SERCA2a were significantly decreased in human failing hearts [214]. Recovery of SUMO-1 conjugation globally or conjugated to SERCA2a improved cardiac function in large animal models [232]. These findings illustrate the participation of SERCA2a sumoylation in human heart failure.
More recently, SENP5 was shown to be implicated in cardiomyopathy and heart failure. SENP5 was upregulated in the human cardiomyopathic heart muscles, and transgenic mice overexpressing human SENP5 specific in hearts recapitulated human cardiomyopathy [233]. The transgenic hearts exhibited increased apoptosis and decreased cell proliferation with enlarged mitochondria. Moreover, overexpression of Bcl2 in the SENP5-Tg hearts rescued cardiac dysfunction [233]. Mechanistically, SUMO conjugation to Drp1, an important factor for mitochondrial fission, was decreased in the SENP5-overexpressing hearts, and knockdown of Drp1 in cultured cardiomyocytes repressed the activation of apoptotic pathway by SENP5 [233]. Thus, SENP5 induces cardiomyopathy mainly via targeting mitochondria.
The abovementioned studies serve as examples demonstrating that deficiency in SUMO conjugation to a particular substrate may contribute to abnormal cardiac muscle disorders. However, the cardiac phenotypes presented in globally sumoylation-deficient murine models should be a consequence of the total or net effect on the activity of multiple SUMO substrates, rather than an effect on the function of a single SUMO target. How to associate the functional consequence originating from a global sumoylation change with a specific group of targets that work in the same or similar signaling pathways is an interesting topic for future investigation.
14.4.2.3 SUMO and Conduction Diseases
Cardiac conduction disorders pose a significant health threat (see Chaps. 62 and 63). The aforementioned sumoylation-deficient lamin A mutants also are associated with familial conduction disease. Another cardiac conduction disease is progressive familial cardiac conduction block I, which exhibits autosomal dominant inheritance [234]. A point mutation from glutamic acid 7 to lysine (E7K) in TRPM4 gene, which encodes transient receptor potential cation channel, subfamily M, member 4 (a Ca2+-activated nonselective cation channel), was responsible for the development of familial heart conduction block [220]. TRPM4 is a SUMO substrate, and the mutant E7K displays a resistance to the desumoylation by SENP1, subsequently protecting it from proteasomal degradation and leading to increased channel activity [235]. SUMO also targets other cardiac ion channel proteins such as Kv2.1 [236] and Kv1.5 [237] and regulates their functions; however, whether sumoylation of these proteins are involved in cardiac arrhythmia needs to be investigated.
14.4.2.4 Potential Implication of SUMO Conjugation in the Ischemic Heart
Several lines of evidence showed that there was an increase in conjugation of SUMO-1 and SUMO-2/3 in cerebral ischemia, which was protective against tissue damage [238–241]. However, no systemic study has yet suggested the involvement of a global change in SUMO conjugation during ischemia/reperfusion. However, there is one study in which a globally increased SUMO-1 conjugation was observed in the hypoxic heart associated with elevated SUMO-1-attached HIF1α [242]. Erk5 (extracellular signal-regulated kinase 5), an important mediator of ischemic/reperfused injury and inhibitor of apoptosis, was suppressed by sumoylation [243]. Sumoylation of Erk5 was increased in myocardial infarction, in H2O2-induced inflammation, and in the aortas of diabetic mice [197, 243]. Although data regarding how sumoylation affects HIF1α stability and function are controversial [188, 244–246], an increase in the level of SUMO-1 conjugated Erk5 appears to promote inflammation, therefore worsening the injury. Given that many SUMO substrates are implicated in ischemic/reperfused heart injury, the exact role the SUMO conjugation pathway plays in this particular pathophysiological setting remains to be determined.
Conclusion
Since the discovery of the first SUMO protein, the number of novel SUMO substrates has increased rapidly, and the understanding of how the SUMO conjugation pathway is involved in a variety of cellular activities as well as the underlying molecular mechanisms has improved significantly. However, to fully decipher the role played by SUMO conjugation in cardiovascular disease, several issues remain to be addressed. For example, is there any functional difference between SUMO-1 and SUMO-2/3 in terms of regulating cardiovascular function? Although a global change in SUMO-1 conjugation was observed in the human failing hearts [214], given the complexity of pathophysiological conditions associated with end-stage heart failure, is this change associated with cardiomyopathy or a consequence of long-term treatment? Also, how SUMO-2/3 functions in the human diseased hearts requires systemic study. Another interesting topic is whether drugs with cardiovascular side effects induce any significant changes in the levels of SUMO conjugation. In yeast, the sumoylation pathway was implicated in doxorubicin-induced cytotoxicity [247]. Whether or not doxorubicin can mediate sumoylation in the heart, which at least partially contributes to doxorubicin-linked cardiomyopathy, is an interesting area to explore.
Acknowledgements
The work from the authors’ lab was supported by the grants from the Texas Higher Education Coordinating Board (THECB), the American Heart Association, and the National Institutes of Health.
References
1.
Stark GR, Wang Y, Lu T (2011) Lysine methylation of promoter-bound transcription factors and relevance to cancer. Cell Res 21:375–380PubMedCentralPubMed
2.
Mostaqul Huq MD, Gupta P, Tsai NP et al (2006) Suppression of receptor interacting protein 140 repressive activity by protein arginine methylation. EMBO J 25:5094–5104PubMedCentralPubMed
3.
Xu W, Chen H, Du K et al (2001) A transcriptional switch mediated by cofactor methylation. Science 294:2507–2511PubMed
4.
Chevillard-Briet M, Trouche D, Vandel L (2002) Control of CBP co-activating activity by arginine methylation. EMBO J 21:5457–5466PubMedCentralPubMed
5.
Paik WK, Cho YB, Frost B et al (1989) Cytochrome c methylation. Biochem Cell Biol 67:602–611PubMed
6.
Sitaramayya A, Wright LS, Siegel FL (1980) Enzymatic methylation of calmodulin in rat brain cytosol. J Biol Chem 255:8894–8900PubMed
7.
Bauer UM, Daujat S, Nielsen SJ et al (2002) Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO Rep 3:39–44PubMedCentralPubMed
8.
Murata K, Kouzarides T, Bannister AJ et al (2010) Histone H3 lysine 4 methylation is associated with the transcriptional reprogramming efficiency of somatic nuclei by oocytes. Epigenetics Chromatin 3:4PubMedCentralPubMed
9.
Nguyen CT, Weisenberger DJ, Velicescu M et al (2002) Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res 62:6456–6461PubMed
10.
Klose RJ, Zhang Y (2007) Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol 8:307–318PubMed
11.
Kaneda R, Takada S, Yamashita Y et al (2009) Genome-wide histone methylation profile for heart failure. Genes Cells 14:69–77PubMed
12.
Pasini D, Bracken AP, Jensen MR et al (2004) Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23:4061–4071PubMedCentralPubMed
13.
Cao R, Zhang Y (2004) SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell 15:57–67PubMed
14.
Shi B, Liang J, Yang X et al (2007) Shang, Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol 27:5105–5119PubMedCentralPubMed
15.
Chen L, Ma Y, Kim EY et al (2012) Conditional ablation of ezh2 in murine hearts reveals its essential roles in endocardial cushion formation, cardiomyocyte proliferation and survival. PLoS One 7, e31005PubMedCentralPubMed
16.
He A, Ma Q, Cao J et al (2012) Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ Res 110:406–415PubMedCentralPubMed
17.
Moskowitz IP, Wang J, Peterson MA et al (2011) Transcription factor genes Smad4 and Gata4 cooperatively regulate cardiac valve development. Proc Natl Acad Sci U S A 108:4006–4011PubMedCentralPubMed
18.
He A, Shen X, Ma Q et al (2012) PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev 26:37–42PubMedCentralPubMed
19.
Sakata Y, Koibuchi N, Xiang F et al (2006) The spectrum of cardiovascular anomalies in CHF1/Hey2 deficient mice reveals roles in endocardial cushion, myocardial and vascular maturation. J Mol Cell Cardiol 40:267–273PubMed
20.
Delgado-Olguin P, Huang Y, Li X et al (2012) Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet 44:343–347PubMedCentralPubMed
21.
Jones B, Su H, Bhat A et al (2008) The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet 4, e1000190PubMedCentralPubMed
22.
Nguyen AT, Xiao B, Neppl RL et al (2011) DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev 25:263–274PubMedCentralPubMed
23.
Lohan J, Culligan K, Ohlendieck K (2005) Deficiency in cardiac dystrophin affects the abundance of the alpha/beta-dystroglycan complex. J Biomed Biotechnol 2005:28–36PubMedCentralPubMed



