Fig. 35.1
Platelets induce chemotactic, adhesive, and proteolytic properties of endothelial cells
However, platelet IL-1β not only modifies endothelial release of chemotactic proteins, but it also has the potential to increase endothelial expression of adhesion molecules. Surface expression of ICAM-1 and αvβ3 on endothelial cells is significantly enhanced by activated platelets via IL-1β [10, 12]. Both enhanced chemokine release and upregulation of endothelial adhesion molecules through platelet-derived IL-1β act in concert and promote neutrophil and monocyte adhesion to the endothelium. IL-1β-dependent expression of early inflammatory genes, such as MCP-1 or ICAM-1, involves the activation of the transcription factor nuclear factor kappa B (NF-κB). Transient adhesion of platelets to the endothelium initiates degradation of IκB and supports activation of NF-κB in endothelial cells, thereby inducing NF-κB-dependent chemokine gene transcription [11]. Parallel to this finding, transfection of “decoy” κB oligonucleotides or a dominant negative IKK mutant attenuates platelet-induced nuclear translocation of NF-κB and MCP-1 secretion in endothelial cells [10]. Likewise, platelet-induced NF-κB-activation was largely reduced by IL-1β antagonists, supporting the notion that platelet IL-1β is the molecular determinant of platelet-dependent activation of the transcription factor. Taken together, platelet-derived IL-1β initiates NF-κB-dependent expression of chemotactic and adhesive proteins in endothelial cells. In this manner, platelets promote the recruitment of both neutrophils and monocytes to the endothelial cell surface, thus inducing inflammation.
Another platelet-derived chemokine is RANTES, which has been identified to trigger monocyte arrest on inflamed and atherosclerotic endothelium [13]. Deposition of platelet RANTES induces monocyte recruitment mediated by P-selectin. Furthermore, release of platelet-derived CD40 ligand induces inflammatory responses in endothelium. CD154 (CD40L), a 30–33 kDa protein, belongs to the TNF family of cytokines, which includes TNF-α and Fas ligand. CD40L was originally thought to be restricted to CD4+ T-lymphocytes, mast cells, and basophils. Henn et al. [14] showed that platelets store CD40L in high amounts and release CD40L within seconds following activation in vitro and in vivo. Ligation of CD40 on endothelial cells by CD40L expressed on the surface of activated platelets increased the release of IL-8 and MCP-1, the principal chemoattractants for neutrophils and monocytes. In addition, platelet CD40L enhanced the expression of endothelial adhesion receptors including E-selectin, VCAM-1, and ICAM-1, all molecules that mediate the attachment of neutrophils, monocytes, and lymphocytes to the inflamed vessel wall (Fig. 35.2). Hence, like IL-1β, CD40L expressed on platelets induces endothelial cells to release chemokines and to express adhesion molecules, thereby generating signals for the recruitment of leukocytes in the process of inflammation. CD40 ligation on endothelial cells, smooth muscle cells, and macrophages initiates the expression and release of matrix degrading enzymes, the matrix metalloproteinases (MMPs). These enzymes, which degrade extracellular matrix proteins, significantly contribute to destruction and remodeling of inflamed tissue. Adhesion of activated platelets to endothelial cells results in generation and secretion of MMP-9 and of the protease receptor uPAR on cultured endothelium [15]. The endothelial release of MMP-9 was dependent on both the fibrinogen receptor GPIIb–IIIa and CD40L because inhibition of either mechanism resulted in reduction of platelet-induced matrix degradation activity of endothelial cells. Moreover, GPIIb–IIIa ligation resulted in substantial release of CD40L in the absence of any further platelet agonist. These results propose that the release of platelet-derived proinflammatory mediators such as CD40L is dependent on GPIIb–IIIa-mediated adhesion. This mechanism may be pathophysiologically important to localize platelet-induced inflammation of the endothelium at a site of platelet-endothelium adhesion.
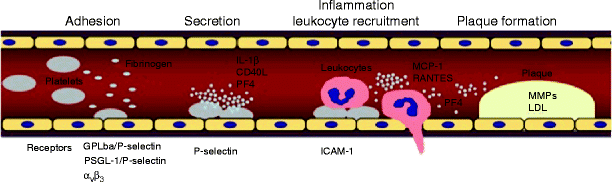
Fig. 35.2
Platelets activate the endothelial monolayer of arteries and induce monocyte chemotaxis and migration, thus propagating atherosclerotic lesion formation
Platelets Interact with Leukocytes
Platelet adhesion to the endothelium or the subendothelial matrix induces platelet activation and the release of substances that are able to cause chemotaxis and migration of circulating leukocytes toward the site of platelet accumulation. Similar to platelet adhesion to the vessel wall, leukocyte recruitment to vascular endothelium requires multistep adhesive and signaling events, including selectin-mediated rolling, leukocyte activation, and integrin-mediated firm adhesion and diapedesis [5]. On leukocytes, members of the β2-integrin family, LFA-1, MAC-1, and p150.95, as well as β1-integrins interact with endothelial counterligands such as ICAM-1, surface-associated fibrinogen [16], or vascular cell adhesion molecule-1 (VCAM-1) to mediate the described heterotypic cell interaction. At sites of platelet adhesion to the endothelium or subendothelium, leukocyte infiltration can occur through interactions with platelets and fibrin [5]. Similar to the leukocyte-endothelium adhesion, a sequential adhesion process of leukocytes to adherent platelets has been proposed. Leukocyte adhesion to platelets involves surface expression of P-selectin on activated platelets and binding to PSGL-1, the counterreceptor present on neutrophils and monocytes [17]. Diacovo et al. [18, 19] have previously demonstrated that leukocytes tether, roll, and subsequently rest on activated platelet monolayers via sequential action of platelet P-selectin and ICAM-2 binding to their leukocyte counterreceptors PSGL-1 and CD11b/CD18, respectively. This suggests that platelets attached to the vessel wall may recruit leukocytes. In addition, P-selectin/PSGL-1-dependent platelet-leukocyte interaction brings platelets into close vicinity with neutrophils and may facilitate leukocyte activation by platelet proinflammatory mediators. Earlier studies indicated that GPIbα and JAM-3 on platelets are potential counterreceptors for MAC-1 [20] and that they mediate mechanism of platelet-leukocyte adhesion. Furthermore, ICAM-2 and αIIbβ3-associated fibrinogen have also been proposed to mediate MAC-1-dependent platelet-leukocyte adhesion. Thus, platelets either immobilized on a surface or activated in suspension express a complete machinery to recruit leukocytes: (1) platelet P-selectin is a mediator of the first contact (tethering), (2) interaction of platelet P-selectin with its counterreceptor PSGL-1 on leukocytes induces signaling events relevant for MAC-1 activation, and (3) the activated β2-integrin (MAC-1) on leukocytes allows and reinforces firm platelet-leukocyte adhesion through binding to counterreceptors (ICAM-2, fibrinogen bound to GPIIb–IIIa, GPIbα, JAM-3) present on the platelet surface.
Platelets Interact with Circulating Progenitor Cells
There is increasing evidence showing that circulating progenitor cells contribute to vascular repair mechanisms and limit atheroprogression. Impairment of progenitor-dependent vascular repair due to either low numbers of circulating progenitor cells or dysfunctional progenitor cells leads to inadequate vascular healing and atheroprogression. Recently, the role of platelets for recruitment and subsequent differentiation of progenitor cells has been recognized [21–23]. Adherent platelets recruit circulating progenitor cells and induce differentiation of the latter into endothelial cells or macrophages and foam cells [21–23]. Further, the combination of platelets and fibrin promoted CD34+ cell migration even to a greater extent than vascular endothelial growth factor in vitro [24]. Moreover, the chemokine stromal cell-derived factor-1 (SDF-1) was found to be secreted by activated platelets, which supports chemotaxis and primary recruitment of progenitor cells on the surface of arterial thrombi in vivo [22, 23]. Moreover, cytokine-mediated deployment of SDF-1 coming from activated platelets induces revascularization through mobilization of CXCR4 hemangiocytes [25].
Adhesion of human CD34+ cells to immobilized platelets is significantly attenuated in the presence of blocking mAbs anti-CD162 or anti-CD62P, indicating that the platelet P-selectin interacts with the endothelial progenitor cells (EPCs) through interaction with P-selectin glycoprotein ligand-1 [24, 26, 27]. Thus, platelets act as an intermediate mediator to tether progenitor cells, indicating that platelets are a prerequisite for the initial step of the homing process of CD34+ cells to vascular injury.
Platelets play a critical part not only in the capture, but also in the subsequent differentiation of murine EPCs, inducing the differentiation of the latter into spindle-shaped cells that are positive for vWF [21]. Furthermore, human CD34 progenitor cells can form colonies on immobilized platelets similar to immobilized fibronectin and further differentiate into mature endothelial cells [23]. Under distinct circumstances, however, in vitro co-culture experiments between platelets and human CD34+ cells induce distinct morphological changes of the latter and differentiation into macrophages at an early phase and later on into foam cells [26].
Inhibition of Platelet Adhesion Attenuates Atheroprogression
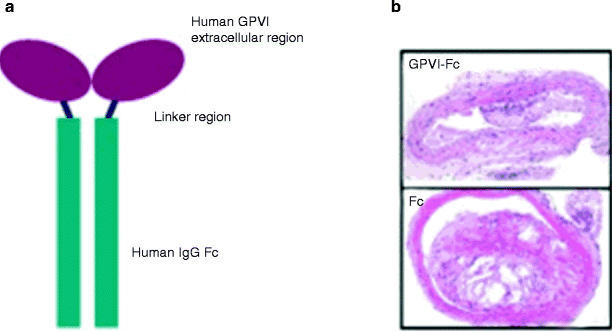
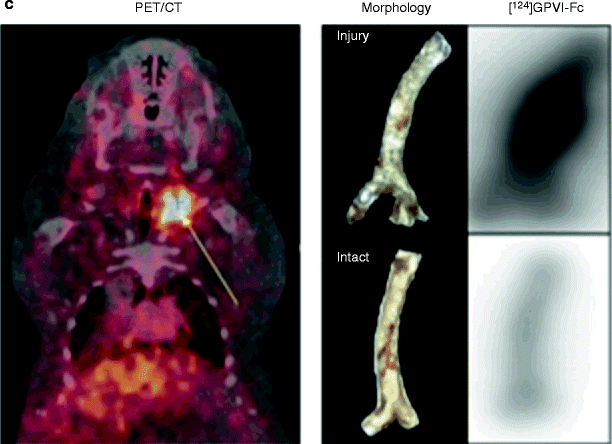
Enhanced chronic interaction of platelets with the arterial wall results in endothelial inflammation and atheroprogression [3]. The platelet von Willebrand receptor GPIbα and the collagen receptor GPVI have been demonstrated to largely contribute to endothelial platelet adhesion in vivo in ApoE −/− mice [28], making them good candidates for inhibition. Inhibition of GP1b prevented adhesion of circulating platelets to endothelial cells at the carotid artery of ApoE −/− mice. Inhibition of platelet adhesion by blocking monoclonal anti-GPIb antibodies substantially attenuated atheroprogression in ApoE −/− mice [28]. Next, we studied the role of GPVI-mediated platelet adhesion for atheroprogression. Prolonged administration of the soluble form of GPVI [28] substantially reduced atheroprogression in ApoE-deficient mice. Further, gene transfer of GPVI-Fc to the carotid vascular wall significantly attenuated atheroprogression and endothelial dysfunction in atherosclerotic rabbits in vivo [29]. In addition, administration of soluble GPVI-Fc preferentially bound to sites of vascular injury and was able to inhibit neointimal formation after wire-induced vascular injury in ApoE −/− mice [30] (Fig. 35.3). Thus, inhibition of platelet adhesion via GPIb- or GPVI-blockers might be a promising strategy to attenuate lesion progression [31].
 < div class='tao-gold-member'>
< div class='tao-gold-member'>




Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
