Plaque Sealing
Bernhard Meier
For quite a while now, we have begun to look at the coronary artery plaque (or any vascular plaque for that matter) as a differentiated entity. Not every plaque means clinical trouble; and when clinical trouble exists, in may range from a potential for myocardial infarction (MI) and death to simply a loss of quality of life. The active or vulnerable plaque represents the former. It is characterized by activated macrophages, a dearth of smooth muscle cells at the surface (hence a thin and brittle cap), and an abundance of inflammatory cells that produce enzymes, such as metalloproteinases, which erode the cap further. The nonobstructive plaque with a thick fibrous cap may be a nuisance, but it is not a real threat. It is characterized by a robust surface with abundant smooth muscle cells and few inflammatory cells or macrophages. It may even harbor a protective potential, because it often represents a scar over a tear in the arterial wall. This scar may be even less rupture prone than the normal endothelium, not unlike the callus after a bone fracture.
A plethora of terms have emerged when dealing with the subject of plaque vulnerability. These terms encompass thin-cap fibroatheroma, plaque erosion, plaque rupture, plaque sealing, plaque passivation, and plaque stabilization. New terms are being coined as this chapter is written. The saying of Thomas Sydenham (1624-1689): “A man is as old as his arteries” (1) has proved true once more. Tiny accidents in coronary or cerebral arteries decide between life and death. They will continue to happen, but it is our goal—and within our reach—to reduce their numbers and push these occurrences into old age. The research, knowledge, and techniques described in this chapter continue to empower us further in the struggle to subdue the vulnerable plaque.
PLAQUE PASSIVATION USING DRUGS
Systemic Administration
Lipid Modification
Plaque regression through statin therapy has been described but remains unconfirmed (2). The fact that statins are of paramount clinical benefit both in primary and secondary prevention points to a multifaceted mode of action. The supposed pleiotropic effects of statins include a reorganization of the plaque, with a relative diminution of the lipid-rich core; deflammation by thickening of the fibrous cap and decrement of foam cells and activated or apoptotic macrophages; enhancement of endothelial function resumption; and antiplatelet activity. Some of these actions are immediate and some take time. Thus, randomized trials of statin efficacy vary in terms of the first appearance of the clinical benefit of statins. Some of these pleiotropic effects, such as the number of macrophages and matrix metalloproteinase-1 expression with reciprocal collagen content increase, have been substantiated in animal trials (3). Some emerge from clinical studies using intravascular ultrasound (IVUS) and variables such as the hyperechogenicity index and plaque volume (4). Finally, some trials were suggested by the otherwise unexplained promptness or extent of clinical benefits suggesting mechanisms beyond cholesterol reduction (5).
Alternative lipid-lowering agents (nicotinic acid, ezetimibe) have yet to produce similar data. However, it can be conjectured that they also lead to plaque passivation via cholesterol lowering and other avenues. Finally, increasing high-density lipoprotein (HDL) cholesterol may, by itself, reduce the risk of plaque rupture. It can be achieved
through the administration of niacin and antioxidant vitamins in addition to statins (6), or simply by vigorous and regular physical exercise.
through the administration of niacin and antioxidant vitamins in addition to statins (6), or simply by vigorous and regular physical exercise.
Antithrombosis
Both superficial plaque erosion and penetrating plaque rupture expose the extracellular matrix to the blood stream. This leads to platelet activation by platelet adhesion molecules and tissue factors. Platelet adhesion leads to platelet aggregation, and this phenomenon goes hand-in-hand with the activation of the clotting cascade, which orchestrates atherothrombosis. It goes without saying that part of this cascade of events can be prevented through the use of antiplatelet agents or anticoagulation. Even once the process has started, it can be stopped through antithrombotic therapy. Whether this represents an additional passivating effect of the vascular wall (7), or whether the effect is limited to the thrombus, remains to be elucidated.
Platelet aggregability can be reduced mainly through four different pathways. For over 100 years, oral acetylsalicylic acid (Aspirin) has been available. It reduces the synthesis of thromboxane A2 from arachidonic acid by blocking cyclo-oxygenase. More recently, thienopyridines, such as ticlopidine and its successor clopidogrel, have been introduced to orally block the adenosin-diphosphate-P2Y12 receptor. The most potent platelet inhibitors are, unfortunately, only available for intravenous use. They are abciximab, eptifibatide, and tirofiban. These agents directly block glycoprotein receptors IIb/IIIa, thus powerfully preventing any of the approximate 100,000 such receptors per platelet from linking with one end of a fibrinogen molecule—the other end of which might then bind to another platelet’s glycoprotein receptor and lead to the permanent connection of the two thrombocytes. Finally, thrombin is a potent platelet activator in addition to being the key element in the coagulation cascade that leads to fibrin clots.
Acetylsalicylic acid has an excellent record for preventing first or subsequent atherothrombotic events in virtually all risk groups (8). Even in the setting of acute MI, in which plaque rupture has already occurred and a thrombus has already formed, acetylsalicylic acid proved beneficial, with an efficacy comparable to that of streptokinase (9). A meta-analysis showed a reduction of mortality from 11.5% to 9.2%, with significant effects also apparent on reinfarction, vascular death, or stroke (8). The effect was comparable with doses of 75 to 1,500 mg, with less bleeding occurring at lower dosages. Additional platelet inhibition can be obtained by adding clopidogrel. The effect starts within hours if a bolus is used or within days without a bolus (10). Recent data in stroke patients even suggest that acetylsalicylic acid may no longer be required once clopidogrel is employed (11). The beneficial effect of clopidogrel also has been corroborated by its supremacy over acetylsalicylic acid alone in randomized trials in acute coronary syndromes. The more conspicuous effect in patients undergoing percutaneous coronary interventions (the ultimate model of plaque rupture) points to its pivotal role in such high-risk situations (12,13). The effect seems to be augmented when clopidogrel treatment is pursued for 1 year, suggesting a secondary prevention in addition to the acute event reduction at the time of initial intervention (14). Based on this finding, some suggest lifelong use of clopidogrel, whereas others consider the benefit not significant enough to be worthwhile (15).
Because the effect of costly intravenous direct glycoprotein IIb/IIIa inhibitors is most notable in the context of an acute intervention or a positive troponin level (16), their role has been relegated to these circumstances and high-risk patients (e.g., those with diabetes). In particular, the anticipated supremacy of abciximab in inhibiting the Mac 1 and vitronectin receptors (instrumental in the apoptosis of vascular smooth muscle cells, induced by the macrophage colony stimulating factor-activated macrophages) (17) has not yet materialized in clinical trials.
The coagulation pathway leading to the red component of the thrombus can be inhibited or modified at several levels (Fig. 16.1). Both internal and external activation of the coagulation pathways merge upon the activation of factor X to factor Xa. The external pathway requires factors V, VII, and IX for this. These factors are vitamin K dependent and can be blocked by warfarin and similar oral drugs (all necessitating monitoring of the international normalized ratio[INR]). Factor Xa can be inhibited by the intravenous drug fondaparinux and partially by low molecular weight heparins (LMWH), which also are applicable subcutaneously. Both require the presence of antithrombin III (AT III), as does unfractionated heparin.
Both heparins exert their main action in thrombin inhibition. The debate as to whether LMWH is superior to unfractionated heparin appears to be put to rest by the Superior Yield of the New Strategy of Enoxaparin, Revascularization, and Glycoprotein IIb/IIIa Inhibitors (SYNERGY) trial (18). It remains to be seen whether new trials will bring out the theoretical advantage of LMWH in terms of a more direct inhibition of factor Xa and reduced risk of thrombocytopenia.
Thrombin inhibition without AT III can be achieved intravenously using bivalirudin or orally by melagatran (i.e., the oral precursor ximelagatran). Thrombin inhibition is the last and most logical point of interference (comparable with glycoprotein IIb/IIIa inhibition in platelet aggregation). Thrombin (also known as factor II) activates the formation of fibrin from fibrinogen. Fibrin can be broken down to its split products by intravenous fibrinolytics as the last resort in plaque passivation that may, however, be too late.
Calcium Antagonists
The capability to reduce lipid oxidation and foam cell formation, together with increased transmembrane calcium transport, attach a prominent antiatherogenic and plaque-smoothing
role to calcium antagonists. For amlodipine, a second-generation dihydropyridine, some clinical benefits have been demonstrated in patients with high cardiovascular risk (19).
role to calcium antagonists. For amlodipine, a second-generation dihydropyridine, some clinical benefits have been demonstrated in patients with high cardiovascular risk (19).
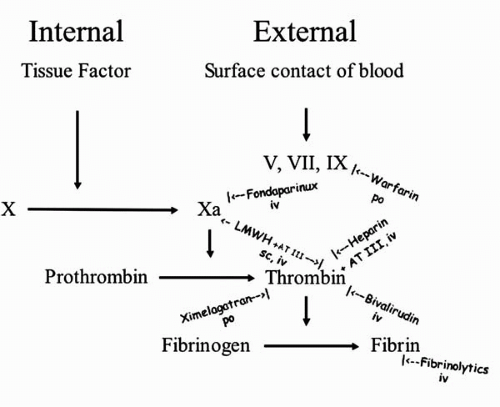 Figure 16.1. Schematic of the coagulation pathway. Roman numerals denote coagulation factors. Iv, intravenous administration; po, oral administration; sc, subcutaneous administration. |
Angiotensin-Converting Enzyme Inhibitors
Angiotensin fosters oxygen free radical production. Its inhibition is therefore likely to improve plaque stability. Moreover, angiotensin-converting enzyme (ACE) inhibitors have been shown to mitigate endothelial dysfunction and decrease macrophage and smooth muscle cell lipoxygenase activity. In general, atherosclerosis was slowed using these compounds in animal models. Clinical trials in high-risk patients and patients with acute coronary syndromes showed benefit from the use of these drugs that appeared more marked than the induced decrement in blood pressure would have predicted (20).
Other Compounds
The highly promising oral antioxidants, in particular some of the vitamins, have emerged as total failures in clinical trials that evaluated atherothrombotic event prevention in various risk groups. Nonetheless, the clear-cut benefit derived from a Mediterranean style diet (21) and the still open debate on the usefulness of a cocktail of vitamin B6, vitamin B9 (folate), and vitamin B12 to lower homocysteine keeps the interest in such compounds alive. Oral immunosuppression using corticosteroids (22) and rapamycin (23) represents a more robust approach to plaque passivation; these agents show some promise without, however, the results of trials being unequivocal.
Local Delivery
The era of catheter-based local drug delivery to premedicate or postmedicate plaques that are on the verge of rupture or have already ruptured has passed, despite the alluring concept of achieving high on-site concentrations without risking systemic side effects. Impediments to its success were the cumbersome techniques used—sweating balloons, enclosing tandem balloons, microneedle injections, and the like—many of which required prolonged episodes of absent coronary flow (7,24). Trying to maintain flow during drug application made the instruments even more complex, expensive, and difficult to handle. Despite a trial showing a reduction of restenosis using local enoxaparin (25), the concept has not been further pursued, not even in conjunction with percutaneous coronary interventions that provide a compelling reason for introducing catheters into the coronary arteries.
In addition, drug-eluting stents (DES) have all but monopolized the attention of interventional cardiologists. This type of local drug delivery is easy to master, necessitates no additional effort, and introduces no added discomfort or risk for the patient. Initially, heparin DES were used, with no apparent advantage (26). Next, DES using dexamethasone showed some benefit in initially unstable patients (27). The dam broke with the first publications of clinical trials using rapamycin (sirolimus) or paclitaxel DES. Although the extent of plaque stabilization was limited to a reduction of restenosis—an annoying yet benign event—DES are set to dominate the arena of coronary stenting. However, the gross mechanical impact of stent
implantation at the lesion site camouflages the plaque passivation effect (if there is any) of these compounds. More interestingly, studies are needed to determine if the eluting drugs stabilize plaques downstream from the DES, at least over a certain time (28).
implantation at the lesion site camouflages the plaque passivation effect (if there is any) of these compounds. More interestingly, studies are needed to determine if the eluting drugs stabilize plaques downstream from the DES, at least over a certain time (28).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


