Fig. 61.1
Changes in MAP in response to chronic reduced uterine perfusion (RUPP) in pregnant rats and following pretreatment with an ETA receptor antagonist (+ETA) in (a) RUPP and (b) normal pregnant rats. *P < 0.05 versus normal pregnant (NP) rats; ‡P < 0.05 versus RUPP rats and NP rats. All data are expressed as mean ± SEM (Adapted with permission from Ref. [29])
61.2.2 Cardiovascular Risk After a Pregnancy Complicated by Placental Insufficiency
Preeclampsia is a multi-organ syndrome that results from the release of soluble factors from the ischemic placenta into the maternal circulation resulting in extensive maternal endothelial dysfunction and hypertension during pregnancy [39]. The long-term risk to develop chronic hypertension [40] and CV disease across the lifespan [41] is increased following a pregnancy complicated by preeclampsia. One potential contributor to this increased lifetime risk may involve the adverse lingering impact of preeclampsia on CV risk factors in the mother such as altered expression of angiogenesis-related proteins [42] and increased markers of metabolic risk including blood pressure, body mass index (BMI), and insulin resistance [43–47]. Blood pressure and proteinuria remain elevated at 6 weeks after a pregnancy complicated by preeclampsia [43]. Circulating levels of SFlt-1 are reported to remain elevated up to 1 year postpartum [46]. A persistent reduction in endothelial function can persist for several years [44], and increases in BMI, insulin resistance, and blood pressure are observed 2–12 years [45] following a preeclamptic pregnancy. Genetics is also thought to contribute to the risk for preeclampsia (for an extensive review, see reference [48]) and may also impact long-term CV risk following a pregnancy complicated by preeclampsia. Thus, numerous studies indicate that preeclampsia increases a women’s CV risk across her lifespan and indicates that her health during pregnancy may provide insight into her future chronic health.
61.3 Low Birth Weight
61.3.1 Etiology of Intrauterine Growth Restriction and Preterm Birth
The etiology of low birth weight includes complications during pregnancy that impact fetal growth including preeclampsia in addition to factors that result in spontaneous preterm delivery. Fetal growth is directly related to nutrient availability, which involves the mother’s nutritional intake in addition to the health of the maternal and uteroplacental circulatory system (for an extensive review, see [3]). Although maternal undernutrition can result in improper growth of the fetus, the main cause of fetal growth restriction within the western world involves placental insufficiency [49]. Placental insufficiency can result in the development of hypertension during pregnancy, and it is well established that birth weight decreases with increasing maternal blood pressure [50]. However, blood pressure is also elevated in normotensive pregnancies complicated by IUGR [51–53] suggesting a common etiology of IUGR related to reductions in uteroplacental blood supply. Vessel morphology including decreased lumen circumference is impaired in pregnancies complicated by IUGR [54, 55]. IUGR is also associated with impaired umbilical vein vascular function [55] and increased resistance within the umbilical artery in association with reduced luminal circumference [54]. Additionally, expression of sFlt-1 is significantly increased [56, 57], and circulating levels of PIGF are decreased in the placenta of normotensive pregnancies complicated by IUGR [57]. Maternal smoking is associated with reductions in uteroplacental perfusion [58], and maternal smoking also increases the risk of a pregnancy complicated by IUGR [59]. Thus, the etiology of IUGR may involve an imbalance in angiogenic and anti-angiogenic factors that leads to impaired uteroplacental perfusion and subsequent IUGR.
Preterm birth is defined as birth before 37 weeks of gestation and is the leading cause of infant morbidity and mortality [60]. The etiology of spontaneous preterm birth involves multiple pathological processes including genetic [61] and environmental factors such as assisted reproductive technology [62], maternal smoking, asthma [59], and maternal stress [63]. Intra-amniotic infections [64] and maternal vascular disease including placental abruption and hemorrhage [65] (for complete review, see reference [60]) are also causative factors for spontaneous preterm delivery. Preterm birth may also be elective due to maternal or fetal complications such as preeclampsia and IUGR [60]. Regardless of these maternal conditions, preterm birth is associated with increased CV risk in the offspring in later life [11].
61.3.2 Developmental Programming of Hypertension
Based on the similar geographical distribution of infant mortality to death from coronary heart disease, David Barker was the first to postulate that factors that slow fetal growth lead to increased CV risk in later life [66]. The inverse association between birth weight and blood pressure supported this original hypothesis [67], and early experimental studies utilizing maternal undernutrition during gestation in the rat to induce hypertension in the offspring provided proof of principle [37, 68]. Currently, it is well established that offspring of pregnancies complicated by preeclampsia [9], preterm birth [11], and IUGR [69] exhibit increased blood pressure in later life. However, despite the etiology of IUGR, slow growth during fetal life is associated with increased blood pressure and CV risk in later life [69]. Extensive investigation into the mechanisms that program increased blood pressure following slow fetal growth indicates that numerous factors contribute to the developmental programming of increased blood pressure and CV risk (for extensive reviews, see [70–72]). The kidney is the primary mediator in the long-term, steady-state control of blood pressure through its ability to regulate chronic fluid and electrolyte balance via the pressure natriuresis mechanism [73]. However, alterations in other regulatory control systems such as the sympathetic nervous system (SNS) [74] and the renin-angiotensin system (RAS) [75] in addition to increased oxidative stress [76] are also indicated in the etiology of essential hypertension (Chaps. 31, 35, and 36). Experimental studies indicate that alterations in these control mechanisms contribute to impaired blood pressure control and the development of hypertension that is programmed in response to slow fetal growth [70–72].
Placental insufficiency in the pregnant rat induces hypertension that is abolished by blockade of the RAS [77] implicating a key role for the RAS in the etiology of hypertension that develops in response to low birth weight. Fetal exposure to a developmental insult leads to significant alterations in expression of the intrarenal RAS in different experimental models of hypertension induced via a development insult (for an extensive review, see reference [10]). In the rat model of IUGR induced via placental ischemia, temporal changes in expression of the intrarenal RAS are observed [78]. The RAS is critical for proper nephrogenesis [79], and a reduction in nephron number is observed in association with hypertension in rat offspring exposed to placental insufficiency [12]. Intrarenal renin and angiotensinogen expression are reduced during nephrogenesis in the neonatal IUGR rat [78], whereas expression of intrarenal renin and angiotensinogen in addition to renal-angiotensin-converting enzyme (ACE) activity is increased in the male IUGR rat in adulthood [78]. These studies suggest that placental ischemia programs a reduction in the intrarenal RAS during nephrogenesis that may contribute to impaired renal development. However, inappropriate activation of the RAS in later life may contribute to the hypertension that develops in offspring following a developmental insult that induces slow fetal growth and low birth weight.
A reduction in nephron number is observed in individuals with low birth weight [80] and is associated with increased risk for early-onset renal failure in low birth weight individuals [81]. Reduced nephron number is also reported in IUGR rat offspring exposed to placental insufficiency [12]. Low birth weight is also associated with increased susceptibility to renal injury in the rat model of IUGR induced by placental insufficiency [82]. Thus, these studies suggest that slow growth during fetal life impairs renal development in a manner that increases renal vulnerability. However, the importance of reduced nephron number in the development of hypertension following slow fetal growth is not entirely clear. Experimental studies indicate that a reduction in nephron number that occurs during nephrogenesis is associated with increased blood pressure in later life [83]. Congenital defects that result in a solitary functioning kidney also lead to hypertension in later life implicating that a nephron deficiency during development can adversely impact the later long-term control of blood pressure [84]. Yet, glomerular filtration rate and effective renal plasma flow are not altered in hypertensive IUGR rat offspring exposed to placental insufficiency [8] suggesting that a reduction in renal function per se leading to a reduction in pressure natriuresis may not be key mediator of IUGR-induced hypertension. However, as discussed above, inappropriate activation of the RAS may contribute to the development of hypertension programmed in response to low birth weight. In addition, the renal nerves may also play a contributory role in the developmental programming of hypertension following fetal exposure to placental ischemia.
Bilateral renal denervation abolishes hypertension in IUGR rat offspring exposed to placental insufficiency [85–87]. However, the development of hypertension in IUGR rat offspring exposed to placental insufficiency is sex and age dependent with male IUGR rats exhibiting hypertension in young adulthood [8] and females developing hypertension in later life [86]. Therefore, these findings indicate that the stimulus for activation of the renal sympathetic nerves in the hypertension that develops in male IUGR prior to puberty and persists into adulthood may differ from the hypertension that develops in female IUGR in later life.
Hypertension in female IUGR rats is associated with marked increases in total fat mass and circulating leptin [86]. Experimental studies indicate that exposure to a high-fat diet leads to increased adiposity associated with an increase in leptin, renal sympathetic nerve activity, and hypertension [88]. Thus, the etiology of hypertension that develops in the female IUGR with age may involve increased adiposity resulting in increased leptin. However, hypertension in the male IUGR develops prior to puberty [8] and is not associated with an increase in adiposity or leptin. Thus, programming of high blood pressure in the male IUGR may be inherent and involve a different mechanism relative to the female IUGR.
One potential mechanism may include activation of the renal sympathetic nerves mediated via central activation of the RAS. Angiotensin II generated within the CV control region of the brain is implicated in the renal control of body fluid balance via activation of the renal sympathetic nerves [89]. Blockade of the RAS via intracerebroventricular administration of a losartan, an AT1 R antagonist, reduces hypertension programmed by maternal protein restriction during fetal life [90] implicating the central RAS in the etiology of hypertension programmed in response to this fetal insult. Although this model of programmed hypertension is induced via maternal undernutrition instead of placental ischemia, systemic blockade of the RAS also abolishes hypertension in low-protein offspring [90, 91], indicating that common mechanistic pathways are involved in the developmental programming of hypertension regardless of fetal insult.
To conclude, these studies indicate that activation of the renal sympathetic nerves contributes to the etiology of hypertension programmed in response to placental ischemia. However, these findings also indicate that the origins of increased renal sympathetic nerve activity may be sex specific implicating that further investigation is needed in order to understand the exact mechanism by which sex impacts the development programming of chronic disease.
Marked increases in renal oxidative stress may also contribute to hypertension induced in response to placental insufficiency. Increases in renal oxidative stress can contribute to renin release and activation of the renal afferent nerves potentiating the development of hypertension [76]. Hypertension in male IUGR rats exposed to placental insufficiency is associated with marked increases in renal markers of oxidative stress [92]. Chronic treatment with tempol abolishes hypertension in male IUGR rats in young adulthood (Fig. 61.2) [92] implicating a role for oxidative stress in the etiology of IUGR-induced hypertension. However, renal markers of oxidative stress are not elevated in normotensive female IUGR rats in young adulthood [92]. Furthermore, renal expression and activity of antioxidant enzymes are increased in female IUGR rats in young adulthood [92] suggesting a compensatory mechanism in the female IUGR.
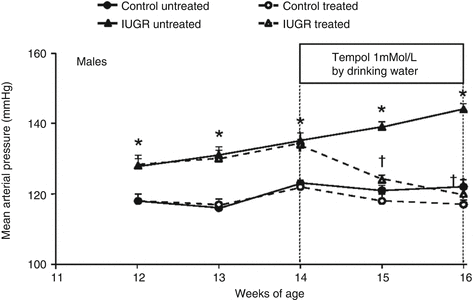
Fig. 61.2
Mean arterial pressure in male control and intrauterine growth-restricted (IUGR) offspring measured by radiotelemetry in conscious, free-moving animals from 12 weeks of age until 16 weeks of age. Animals received the superoxide dismutase (SOD) mimetic tempol (1 mmol/L) or vehicle (tap water ad libitum) for 2 weeks (14–16 weeks of age). *P < 0.05 versus control-treated and untreated offspring. †P < 0.05 versus untreated counterpart. Data values represent mean ± SEM (Adapted with permission from Ref. [92])
Clinical studies indicate that the risk for preeclampsia is increased in low birth weight women [93, 94]. The mechanisms that contribute to increased risk for preeclampsia in a woman born with low birth weight are not yet clear. Risk factors for preeclampsia include endothelial dysfunction [95] and a history of chronic conditions such a hypertension, type 2 diabetes, and kidney disease. Low birth weight is associated with endothelial dysfunction [96]. Low birth weight also increases the risk for type 2 diabetes [97], hypertension [67], and renal disease [81] suggesting that low birth weight programs risk factors that enhance susceptibility to preeclampsia. Genetics may also be a contributory factor [98].
Therefore, experimental studies are providing insight into the etiology of hypertension that develops in the offspring following a pregnancy complicated by preeclampsia and/or low birth weight. Yet, the translation of the impact of low birth weight and gestational history in consideration of therapeutic interventions and treatments for blood pressure control in later life of an individual born with low birth weight or from a pregnancy complicated by preeclampsia is not yet considered. Additionally, findings from experimental studies indicate that males and females differ in their response to slow fetal growth and highlight the importance of further investigation to discern how sex impacts chronic health following a developmental insult.
61.4 Pharmacological Management of Hypertension During Pregnancy
Clinical trials for the treatment of hypertension in pregnancy are limited, and the use of antihypertensive agents during pregnancy is associated with greater risk for adverse outcomes including IUGR and preterm delivery [99]. However, a task force on hypertension in pregnancy recently provided recommendations for the treatment and management of preeclampsia [6]. The use of antenatal steroids was recommended to improve pulmonary function in preterm babies, whereas magnesium sulfate was indicated for women with severe preeclampsia, eclampsia, or HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome [6]. The use of antioxidants and vitamins C and E to prevent preeclampsia was contraindicated, and caution was indicated for the use pharmaceutical approaches that inhibited pathways of the RAS including ACE inhibitors and ARBs [6] (Chap. 36). Labetalol, nifedipine, and methyldopa were suggested for the early management of hypertension, and the use of daily low-dose aspirin was recommended to prevent the development of preeclampsia in women at risk [6]. Clearly additional studies are needed to ensure a benefit to maternal outcome versus an adverse impact on fetal development in the pharmaceutical management of a pregnancy complicated by preeclampsia. Investigation into best practices for the management of hypertension in low birth weight individuals is very limited. A survey of antihypertensive medications in Medicaid recipients noted a greater use of calcium channel antagonists in low birth weight black women, whereas ACE inhibitors were more likely prescribed for low birth weight white men [100]. These findings are suggestive of potential insight into physiological differences that may contribute to racial disparities in hypertension management. However, additional studies are warranted to determine the most effective therapy for low birth weight individuals.
61.5 Concluding Remarks
Placental ischemia during pregnancy serves as the initiating event in the etiology of preeclampsia. Placental ischemia impacts fetal growth and development and also exerts a long-term adverse impact on the CV health of the mother and her child. The risk of preeclampsia is increased in women born with low birth weight implicating the transgenerational effect that placental insufficiency in one generation can have on the CV and gestational health of subsequent generations. Clearly the identification of markers for early diagnosis and the development of therapeutic interventions to prevent the development of preeclampsia and low birth weight are warranted to improve chronic health outcomes in women across their lifespan and that of their children.
References
1.
Avagliano L, Garò C, Marconi AM. Placental amino acids transport in intrauterine growth restriction. J Pregnancy. 2012; 2012: 972562
2.
Brett KE, Ferraro ZM, Yockell-Lelievre J, Gruslin A, Adamo KB. Maternal-fetal nutrient transport in pregnancy pathologies: the role of the placenta. Int J Mol Sci. 2014;15:16153–85.CrossRefPubMedCentralPubMed
3.
4.
5.
Ladyman SR, Augustine RA, Grattan DR. Hormone interactions regulating energy balance during pregnancy. J Neuroendocrinol. 2010;22:805–17.PubMed
6.
American College of Obstetricians and Gynecologists, Task Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet Gynecol. 2013;122:1122–31.
7.
Backes CH, Markham K, Moorehead P, Cordero L, Nankervis CA, Giannone PJ. Maternal preeclampsia and neonatal outcomes. J Pregnancy. 2011;2011:214365.CrossRefPubMedCentralPubMed
8.
9.
10.
11.
de Jong F, Monuteaux MC, van Elburg RM, Gillman MW, Belfort MB. Systematic review and meta-analysis of preterm birth and later systolic blood pressure. Hypertension. 2012;59:226–34.CrossRefPubMedCentralPubMed
12.
Wlodek ME, Westcott K, Siebel AL, Owens JA, Moritz KM. Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int. 2008;74:187–95.CrossRefPubMed
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


