Fig. 2.1
Possible toxic effects of chemotherapy and their relationship
The first is the induction of arterial hypertension, mainly due to angiogenesis inhibitors. The occurrence of hypertension in a patient with underlying myocardial dysfunction acts as a stressor and a possible precipitating factor of heart failure and may also induce other complications in other systems and organs susceptible to very high values of arterial pressure.
Pro-coagulation condition is common in subjects with malignancies. Many chemotherapy agents may enhance this state leading to venous and arterial thromboembolism and clinical occurrence of pulmonary embolism or peripheral venous or arterial thrombosis.
Partially linked with the coagulation altered condition, partially for direct induction can be observed myocardial ischemia and myocardial infarction.
The coagulation problems and the coronary disease treatment require the solution of difficult issues in a patient with malignancy, like the necessity of anticoagulant agents or the insertion of coronary stents.
Finally many chemotherapy agents can induce a prolongation of the ventricular depolarization and of the QT period or arrhythmias with different mechanisms.
This situation in a patient otherwise particularly sensitive to arrhythmias for intrinsic conditions, like electrolyte disturbances or metabolic impairment, can induce harmful situations and also sudden death. In these situations it is mandatory to enclose in the follow-up a periodic evaluation of heart rhythm and a punctual correction of proarrhythmic conditions.
Box 2.1: Physiopathology and Toxic Heart Effects of Chemotherapy Drugs (General Principle)
Type I toxicity (cell necrosis – permanent cardiac damage)
Type II toxicity (cell dysfunction – reversible cardiac damage)
Potentiation of toxic effect by incorrect timing of association of type I and II drugs
Shift from concentrated high doses of chemotherapy in advanced stages of cancer to modulated chemotherapy with combinations of different agents (type I and II), lower doses, and prolonged administration
Shift from prolonging survival to side effects and QoL
Older patients
Not only heart failure
2.3 Type I Agents
2.3.1 Anthracyclines
The first studies on antibiotics anticancer activity were done in the late 1950s, and the first description of cardiac toxicity of anthracyclines came in 1967. From that time the research to discover new toxic drugs began.
Toxicity is related to many different factors: (1) total dose, (2) age (younger and older ages), (3) individual genetic predisposition (probably of genes regarding protective pathways against oxidative damage and transport mechanisms of the drugs), (4) individual sensibility due to preexisting cardiovascular diseases (hypertension, left ventricular dysfunction for multiple causes), and (5) contemporary or previous use of radiotherapy and/or of other chemotherapy drugs (cyclophosphamide, trastuzumab, paclitaxel).
The histopathological damages are similar to that of a typical dilated cardiomyopathy, with interstitial fibrosis, vacuolated myocytes (Adria cells) with loss of myofibrils, disorganization of chromosomes, and infiltration of inflammatory cells.
The main toxicity mechanisms of anthracyclines include the generation of ROS through the formation of iron complexes in the mitochondria, a different kind of interaction with DNA leading to inhibition of its synthesis and inhibition of topoisomerase leading to apoptosis but also to mitochondrial dysfunction, calcium overload, protein degradation, and SERCA dysfunction with sarcomere disruption; moreover, a reduction in antioxidant activity and stem cell production, titan disruption, and altered expression of genes of structural proteins have been shown.
Clinical occurrence of toxicity may be acute (1 day to 1 week), early chronic (1 week to 1 year), or late chronic progressive (1 year to >20 year). The acute phase occurs in 1–11 % of patients, often may be asymptomatic and neglected, only showed by troponin increase or by bioptic specimens. Sometimes patients may suffer chest pain and many kinds of arrhythmias with a prolongation of QT interval at the ECG. The early chronic phase and the late chronic phase are characterized by an LVEF reduction initially without symptoms and then leading to the classic heart failure symptoms and signs. Chronic toxicity may occur in 2 % of treated at 2 years to 5 % at 15 years. These percentages are more elevated in childhood cancer survivors. When overt heart failure is present, the prognosis is very poor with a short-term mortality of 50 %.
The diagnosis of toxicity requires a deep anamnestic collection and physical examination, an ECG, and a chest X-ray, BNP, and troponin evaluation, but all these tests are not specific. Biopsy may instead show the typical Adria cells of anthracycline damage.
Different imaging methods like ECHO, RNA, CMR, and CT may be used to assess LVEF, everyone with pros and contras (radiation exposure, poor echo window); it is essential however to maintain the same method for Follow-Up (F-U) evaluations to compare the results. F-U timing for asymptomatic patients is still a matter of debate: it seems reasonable to undertake controls of ventricular function after the conclusion of chemotherapy at 6 months, 1 year for the following 3 years, and then every 3–5 years lifelong.
Different drugs have been used for toxicity prevention: beta-blocker agents (carvedilol), RAAS inhibitors (enalapril, valsartan), more recently ranolazine, and mainly antioxidant agents like the iron-chelating agents dexrazoxane. Also vincristine in association may reduce toxicity of anthracyclines. On the other end, a different approach has been the modification of pharmacokinetic characteristics to modify uptake and release to the myocardium of the anthracycline. However only a right prevention strategy based on tight interaction between the oncologist and cardiologist may reduce the risk of toxicity of the patient. For every situation a correct risk on benefit ratio has to be calculated: if the subject has no cardiovascular risk factors and may have a great advantage by anthracyclines, he/she must be treated, and if not, he/she must not receive this drug class. A particular attention has to be made to the intermediate category, in which a tight clinical and instrumental control to diagnose initial cardiac damage and to evaluate the possibility to continue the therapy has to be undertaken. Unfortunately only one-third of patients receive preventive measures if asymptomatic LVEF reduction develops, and only half receive a cardiac consultation. The treatment of overt heart failure in patients with anthracycline toxicity is the same of other causes of heart failure.
2.3.1.1 Introduction
The idea to find anticancer antibiotics from soil-based microbes appeared in the 1950s.
An antibiotic isolated from a new strain of Streptomyces peucetius was shown to have a good activity against mouse tumor. Clinical trials began in the 1960s, and the drug was demonstrated to be successful in treating acute leukemia and lymphoma.
In 1967 it was described probably for the first time that daunorubicin could produce fatal cardiac toxicity [6]. Later it was discovered that minor changes in the structure of the compound could lead to changes in biological activity limiting this toxicity. A strain of mutated Streptomyces produced a different antibiotic named Adriamycin; the name was later changed into doxorubicin in adaptation to the established naming convention. Doxorubicin showed also a better activity than daunorubicin against mouse tumors and especially solid tumors.
Although the higher therapeutic index compared to that of daunorubicin, cardiotoxicity was still present.
Daunorubicin and doxorubicin are the prototypes for the anthracycline family. This drug category has now over 2,000 known analogues of doxorubicin.
Anthracyclines and the analogue non-anthracycline mitoxantrone are still among the most effective and used chemotherapeutic agents, mainly for solid tumors, like breast cancer and also for lymphomas, leukemia, and sarcomas.
2.3.1.2 Dose Relationship and Risk Factors for Cardiac Toxicity of Anthracyclines
There is a typical correlation between cardiac toxicity and the cumulative dose of the drug administered.
Patients with no generic risk factors or cardiac risk factors can tolerate doses of doxorubicin up to 300 mg/m2, with a heart failure incidence of less than 2 % [12]. At a cumulative dosage of 400–450 mg/m2, the expected occurrence of heart failure increases to 5 %. The incidence becomes 18 % when the dose is 551–600 mg/m2 and 36 % when the dose exceeds 600 mg/m2 [13].
Following these observations the doses considered at present to reduce the cumulative toxicity are 240–360 mg/m2 of doxorubicin and 450–600 mg/m2 of epirubicin with an expected risk of heart failure of 2–3 % during a period longer than 5 years.
Cancer therapy in childhood and adolescence predisposes to the development of doxorubicin cardiomyopathy in adults. Age influences the risk of developing doxorubicin cardiomyopathy, and very young or very old individuals are more subject to this complication. Children or patients older than 65 years may develop heart failure in a percentage as high as 10 % also at inferior doses [12–14].
In children anthracycline toxicity may play a particular role due to the incomplete cardiac development and interfere with the heart maturation, although the mechanisms of this action are not still known. Cardioprotection and strict surveillance are mandatory in a pediatric population [14]
The individual sensitivity to cardiac toxicity of anthracyclines is largely variable.
Probably there are genetic conditions that predispose to the development of anthracycline-induced cardiotoxicity in some patients, and there is an increasing interest in identifying gene polymorphisms associated with a greater sensitivity to the cardiotoxic effects of anthracyclines. In a study of patients with non-Hodgkin’s lymphoma, the evaluation of single-nucleotide polymorphisms in 82 candidate genes hypothesized to be associated with the development of anthracycline cardiotoxicity [8–11, 15] and identified polymorphisms in genes encoding three proteins: NAPD (H) oxidase, implicated in reactive oxygen species generation, and the doxorubicin efflux transporters MRP1 and MRP2.
In survivors of high-risk childhood ALL, the risk of doxorubicin-associated myocardial damage was particularly elevated in the patients with a C282Y mutation, associated with hereditary hemochromatosis [60].
However, while these studies have focused on specific genes, no genome-wide association studies have been proposed to determine other genes that may identify individuals at increased risk of anthracycline cardiotoxicity.
A risk factor to develop cardiac toxicity is a history of cardiovascular disease such as hypertension and reduced LV ejection fraction before therapy.
Arterial hypertension, previous cardiovascular pathology, and the combination of the treatment with thoracic radiotherapy or with alkylating or anti-microtubule drugs, particularly concomitant treatment with cyclophosphamide, trastuzumab, or paclitaxel, increase the cardiac toxicity of anthracycline (Table 2.1). However every condition that leads to a generic increased myocardial susceptibility to external injuries or that decreases the possibility of recovery of myocardial cells may act in combination with anthracycline to enhance their effects [28].
Table 2.1
Cardiotoxicity of chemotherapeutic agents
Drug class/name | Cardiac adverse effects | Frequency | Note |
|---|---|---|---|
Type I agents | |||
Anthracyclines/anthraquinolones | |||
Doxorubicin | CHF/LV dysfunction | Frequent | LV dysfunction secondary to free radical production and multiple mechanisms; risk depending on cumulative dose and schedule, age, radiotherapy, female gender, previous cardiac disease; continuous infusion, can reduce toxicity. Liposomal delivery systems; dexrazoxane |
Daunorubicin | |||
Epirubicin, idarubicin | |||
Mitoxantrone | CHF/LV dysfunction | Relatively frequent | Acute myocarditis and arrhythmia during infusion |
Alkylating agents | |||
Busulfan | Endomyocardial fibrosis | Uncommon | |
Cardiac tamponade | Uncommon | ||
Cisplatin | Ischemia | Relatively frequent | |
Hypertension | Very frequent | ||
CHF | Relatively frequent | Risk depending on age, radiotherapy, prior anthracyclines | |
Cyclophosphamide | Pericarditis/myocarditis | Frequent | Hemorrhagic myocarditis, at high doses |
CHF | Risk depending on cumulative dose, age, radiotherapy, prior anthracyclines | ||
Ifosfamide | CHF | Relatively frequent | Risk depending on cumulative dose, prior anthracyclines |
Arrhythmias | Relatively frequent | ||
Mitomycin | CHF | Relatively frequent | Risk depending on cumulative dose, age, radiotherapy, prior anthracyclines |
Antimetabolites | |||
Capecitabine | Ischemia | Uncommon | More common in previous CAD; mechanism probably vasospasm or thrombosis |
Cytarabine, Ara-C | Pericarditis | Uncommon | Rare cardiomyopathy after high dose/combination with cyclophosphamide |
CHF | Uncommon | ||
Fluorouracil | Ischemia | Relatively frequent | Risk depending on CAD, radiotherapy, cisplatin, rate and dose; possible mechanism vasospasm |
Cardiogenic shock | Uncommon | ||
Anti-microtubule | |||
Paclitaxel | Sinus bradycardia, AV block, ventricular tachycardia | Uncommon | |
Hypotension | Uncommon | Hypersensitivity; CHF possible with doxorubicin | |
CHF | Relatively frequent | ||
Vinca alkaloids | Ischemia | Relatively frequent | Risk depending on CAD or radiotherapy |
Type II agents | |||
Biological agents | |||
Monoclonal antibodies | |||
Alemtuzumab | Hypotension | Frequent | Infusion reactions |
CHF | Uncommon | ||
Bevacizumab | Hypertension | Frequent | Severe hypertension (>200/110 mmHg) frequent complications |
CHF | Relatively frequent | Concurrent anthracyclines | |
DVT | Uncommon | ||
Cetuximab | Hypotension | Uncommon | Severe infusion reactions (bronchospasm, stridor, urticaria) |
Rituximab | Hypotension | Relatively frequent | Infusion reactions (hypotension, hypoxia, bronchospasm) |
Angioedema | |||
Arrhythmias | Relatively frequent | Rare fatal cardiac failure | |
Trastuzumab | CHF/LV dysfunction | Relatively frequent | LV dysfunction uncommon as single agent, risk dependent on concomitant cyclophosphamide, anthracyclines, and/or paclitaxel |
Interleukins | |||
IL-2 | Hypotension | Very frequent | Higher doses, with vascular leak syndrome (hypotension, hypoperfusion, edema, and effusions); transient LV dysfunction during infusion |
Arrhythmias | Relatively frequent | ||
Denileukin diftitox | Hypotension | Very frequent | With vascular leak syndrome (hypotension, edema, hypoalbuminemia) |
Interferon-α | Hypotension | Frequent | Risk depending on preexisting cardiac dysfunction or prior cardiotoxic therapy |
Ischemia | Relatively frequent | ||
LV dysfunction | Uncommon | ||
Miscellaneous | |||
All-trans retinoic acid | CHF hypotension | Relatively frequent | With retinoic acid syndrome (respiratory distress, fever, pulmonary infiltrates) |
Pericardial effusion | Relatively frequent | ||
Uncommon | |||
Arsenic trioxide | QT prolongation | Very frequent | Monitor QTc and electrolytes – discontinue QT-prolonging drugs |
Imatinib | Pericardial effusion, | Relatively frequent | |
CHF edema | Frequent | Dose related (>300 mg/day) | |
Pentostatin | CHF | Relatively frequent | After high-dose cyclophosphamide before bone marrow transplantation |
Thalidomide | Edema | Relatively frequent | |
Hypotension | Uncommon | ||
DVT | Relatively frequent | ||
Bradycardia | Relatively frequent | ||
Etoposide | Hypotension | Relatively frequent | During rapid infusion |
In comparison with patients treated with other chemotherapeutic agents, patients receiving anthracycline are five times more exposed to the risk of developing heart failure [16].
The interaction between anthracyclines, such as doxorubicin and trastuzumab, is of particular interest given the relatively common use of this agent for adjuvant therapy for breast cancer. A recent review of two large trials comparing the use of chemotherapy with doxorubicin and cyclophosphamide alone with the use of the two agents in addition to an adjuvant treatment with trastuzumab demonstrated an incidence of congestive heart failure of 0.45 % for chemotherapy alone versus 2.0 % for chemotherapy plus adjuvant trastuzumab [48].
The specific problem of the association of anthracyclines with other chemotherapy agents like trastuzumab will be discussed later.
Box 2.2: Factors Associated with Increased Risk of Anthracycline-Induced Cardiotoxicity
Age >65 years or <4 years
Female gender
Hypertension
Preexisting cardiac disease
Mediastinal irradiation
Treatment with cyclophosphamide, paclitaxel, or trastuzumab
Cumulative anthracycline dose
Higher individual anthracycline doses
2.3.1.3 Cardiac Morphology and Histopathological Modifications in Anthracycline Toxicity
Histopathological changes have been shown even in endomyocardial biopsy specimens from patients who have received a dose of doxorubicin as little as 240 mg/m2. The cardiac morphologic and functional modifications of doxorubicin cardiomyopathy are similar to those of dilated cardiomyopathy. In doxorubicin cardiomyopathy there are areas of patchy myocardial interstitial fibrosis and scattered vacuolated cardiomyocytes (Adria cells; Figs. 2.2 and 2.3 – Ref. [20]). Essential expression of doxorubicin cardiotoxicity is myocyte vacuolar degeneration with partial or total loss of myofibrils (Fig. 2.3 – Ref. [20]). Remnants of Z discs can be seen. Distention of the sarcoplasmic reticulum and the T tubules can be present. Coalescence of myocyte vacuoles forms large membrane-bound spaces. The nucleus–chromatin disorganization and replacement of chromatin by pale filaments are also features of doxorubicin cardiomyopathy [20, 25–27].
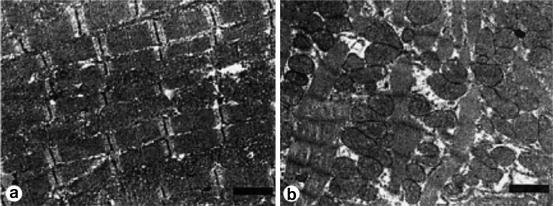
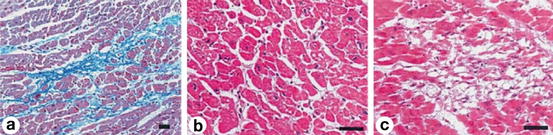
Fig. 2.2
(a) Normal myocyte structure and without abnormal interstitial fibrosis. (b) Myofibrillar loss and vacuolization (Adria cells) and extensive diffuse fibrosis (Published with permission from Takemura et al.)
Fig. 2.3
(a) Normal myocardium with little or no extracellular matrix changes and intact myocytes. (b) Same features in higher magnification. (c) Myocyte loss, matrix disorganization, and diffuse fibrosis (Published with permission from Takemura et al.)
Areas of acute myocyte damage are however not frequent.
Areas of fibrosis are usually widespread, and histiocytic infiltration and fibroblast proliferation are present in the areas of healed myocarditis.
Contractile function and ventricular ejection fraction are reduced, while all cardiac chambers may be dilated although not so severe than that observed in ischemic and nonischemic dilated cardiomyopathies. Concomitant diastolic dysfunction is present. Wall stress is increased due to insignificant change in LV wall thickness. In some patients mural thrombi can be present.
2.3.1.4 Mechanism of Anthracycline Toxicity
The anthracycline-induced toxic mechanism on cardiac cells is very complex and not fully explained at a cellular level.
The first step of the process is the entry of the drug into the myocardial cell.
The mechanisms of the therapeutic effects of doxorubicin on cancer cells are probably different from those responsible for its cardiotoxic mechanisms. These mechanisms of action proposed for the antitumor effects include generation of reactive oxygen species, intercalation into DNA leading to inhibition of synthesis of macromolecules, DNA binding, and DNA cross-linking; furthermore is involved the DNA damage obtained by inhibition and/or poisoning of topoisomerase IIB, and induction of apoptosis by inhibition of topoisomerase II [20, 25, 26]. The principal mechanism proposed for doxorubicin cardiotoxicity is increased oxidative stress, as evident from increased levels of reactive oxygen species (ROS) and lipid peroxidation [20]. Doxorubicin induces toxic damage of the myocardial mitochondria.
Inside the cell the first mechanism is the formation of iron complexes and the production of reactive oxygen species (ROS) that cause impairment in mitochondrial function.
Several mitochondrial enzymes such as NADH dehydrogenase, cytochrome P-450 reductase, and xanthine oxidase are involved in generating oxygen free radicals (ROS). Doxorubicin also increases superoxide formation by increasing endothelial nitric oxide synthase.
The improvement of the cardiotoxic effects of doxorubicin has further demonstrated in transgenic mouse models the importance of modulating reactive oxygen species production. Overexpression of manganese-dependent superoxide dismutase (Mn-SOD) decreases apoptosis and improves left ventricular function in mice treated with doxorubicin, while deletion of Mn-SOD enhances the cardiotoxic effects of the agent [33, 34]. It has demonstrated that doxorubicin metabolism in the cardiomyocytes involves activation of the transcription factor, aryl hydrocarbon receptor, which increases expression of drug-metabolizing proteins [35]. Deletion of aryl hydrocarbon receptor from mice results in increased cardiomyocyte reactive oxygen species generation and apoptosis in response to doxorubicin treatment as well as increased left ventricular dysfunction [36].
The ROS produced by doxorubicin metabolism in cardiomyocytes can subsequently cause cell death through apoptotic pathways (Fig. 2.4) [36–38], started by caspase 9 and caspase 3 activation [39, 40], that lead to the opening of the mitochondrial permeability transition pore and to subsequent release of cytochrome C into the cytosol [41, 42]. Doxorubicin can directly bind cardiolipin, disrupting the association of inner mitochondrial membrane proteins with it; this action could enhance cytochrome C release in response to oxidant stress.
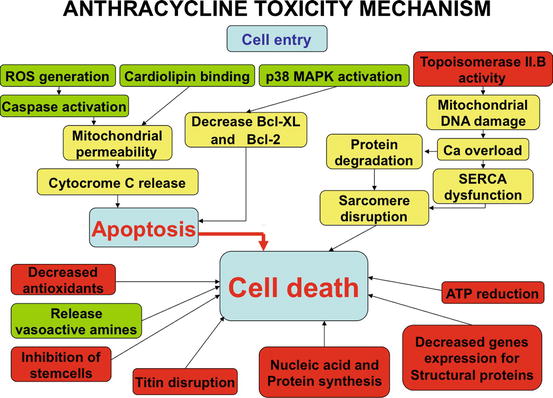
Fig. 2.4
Cellular mechanisms responsible for increased apoptosis and death in cardiac myocytes by doxorubicin. In red inhibitory mechanisms, in green activation mechanisms
Another mitochondrial damage mechanism of doxorubicin can be against the mitochondria genome by interfering with topoisomerase IIβ activity [61]. This ATP-dependent nuclear enzyme is important in regulating the breakage-and-reunion process during replication of mitochondrial DNA. Doxorubicin stabilizes the topoisomerase II–DNA complexes and thereby prevents reassembly of the breaks in DNA strands, an action that can compromise the synthesis of mitochondrial DNA-encoded respiratory chain units [62].
Apoptosis by doxorubicin can be activated also in a different path and mediated in part by p38 MAPK activation. Early after doxorubicin exposure there is evidence that there is an initial upregulation of the expression of the antiapoptotic proteins, Bcl-XL and Bcl-2, followed by decreases in their expression; increasing the expression of Bcl-XL or Bcl-2 could protect against doxorubicin-induced cardiotoxicity.
Cardiomyocyte death is determined by the balance between cytotoxic pathways and cytoprotective pathways. The comprehension of these cytoprotective pathways may provide insights into new possibilities of decreasing the toxicity of anthracyclines.
Antioxidant treatment can cause Akt activation [43–45], and it has been shown that increasing Akt activity through adenoviral vector delivery of constitutively active Akt improves left ventricular function following treatment with doxorubicin. Furthermore, the protective effects of Akt activation include the inhibition of apoptosis, through the inactivation of caspase 9 and caspase 3. In addition, Akt activation is associated with increased expression of the antiapoptotic protein, Bcl-2 [46, 47].
Of clinical interest, one study has demonstrated that neuregulin-1β, the ligand of the epidermal receptor kinase, erbB2, activates Akt in adult rat ventricular myocytes and decreases myocyte disarray [3]. ErbB2 is the target of trastuzumab, which is thought to inactivate erbB2 receptor signaling in breast cancer cells, and has been shown to also cause left ventricular dysfunction. Furthermore, deletion of proteins that can stimulate the production of free radicals by doxorubicin can prevent the decline in left ventricular function following the treatment.
The mitochondrial failure leads to alterations in calcium homeostasis and consequently to the impairment of the contractile function.
Mitochondrial damage could explain the initial finding of diastolic dysfunction in cardiac toxicity by anthracycline [13]. Increasing the dosages can be the cause of a SERCA dysfunction due to interference with the ryanodine receptors of the sarcoplasmic reticulum with a calcium overload that can produce protein degradation and disruption of contractile proteins and of the sarcomere.
Further elevation of the doses causes cell apoptosis and death.
Other proposed mechanisms [20] are decreased levels of antioxidants and sulfhydryl groups, inhibition of nucleic acid and protein synthesis, release of vasoactive amines, altered adrenergic function, and decreased expression of cardiac-specific genes. The downregulation by doxorubicin of genes for the synthesis of proteins like α-actin, myosin light and heavy chains, troponin-I, and desmin has been suggested as a potential mechanism of its cardiotoxicity.
A reduced production of the contractile proteins is associated with myofibrillar loss and depression of myocardial contractile function. Abnormal diastolic function may be caused by downregulation of sarcoplasmic reticular ATPase.
It has been suggested that doxorubicin can inactivate extracellular signal-regulated kinase (ERK). Moreover there is evidence that doxorubicin induces apoptosis of cardiomyocytes thorough the formation of hydrogen peroxide and superoxide thorough the induction of p53 and activated p53 that promote apoptosis of cardiomyocytes.
Endomyocardial biopsies performed at this stage confirm the cell disruption and loss [19]. Several other actions ascribed to doxorubicin could contribute to myocyte dysfunction, including transcriptional changes in intracellular ATP, downregulation of sarcoplasmic reticulum calcium–ATP messenger RNA expression, suppression of transcription factors that regulate cell survival and synthesis of sarcomeric proteins, and disruption of the sarcomeric protein, titin [63].
At the end of this description, another possible negative effect could be mediated by inhibition of myocardium stem cells, endogenous myocardium cells (progenitor cells), that can differentiate into cardiac contractile myocytes [64]. These cells participate in myocardial growth during adolescence and provide a reserve for replacing injured cells in the adult heart. The reduction of these progenitor cells caused by the toxic action of doxorubicin, and of consequent regenerative possibility, could further predispose the heart to contractile dysfunction.
So in conclusion much more than one mechanism is involved in creating myocardial dysfunction by anthracyclines, but the role of many of these remains undefined.
The underlying cause of the electrophysiological cardiotoxicity is unknown, although the efficacy of dexrazoxane in attenuating this injury suggests that the toxicity may be mediated through free radical generation.
2.3.1.5 Clinical Expressions of Anthracycline Toxicity
Anthracycline cardiotoxicity can arise according to the time of occurrence of signs or symptoms of toxicity in three distinct modalities (acute, early onset, and late onset chronic progressive), in relationship to the period of time elapsed from treatment, speed of their development, and speed of worsening of heart failure.
The cardiac damage occurs in the first stages of exposition to the drug immediately or early after administration of an anthracycline. This drug action is demonstrated by the findings coming from endomyocardial biopsies and from the elevation of troponin I soon after the treatment [17, 18].
The clinical effects in these initial times after the administration of an anthracycline may be completely neglected and not perceived by the patient and the clinician.
The incidence of acute cardiotoxicity is considered, according to different studies, between <1 and 11 % [7, 19] occurring during and within 2–3 days of its administration, usually not later than a week from the infusion.
The clinical expressions are usually chest pain due to myopericarditis and/or palpitations due to sinus tachycardia, paroxysmal nonsustained supraventricular tachycardia and premature atrial and ventricular beats, and sinus node dysfunction. The electrocardiogram may show nonspecific ST–T changes, left axis deviation, and decreased amplitude of QRS complexes and ventricular late potentials. All anthracyclines can prolong corrected QT (QTc) intervals measured on surface electrocardiography [65].
The mechanisms for these acute changes are not clear but may be due to doxorubicin-induced myocardial reversible edema [19, 20] that can follow to an acute inflammatory response, which differs from the generally accepted cause of the chronic anthracycline cardiotoxicity discussed above.
A recent case report suggested that treatment with anthracyclines may also produce a stress-induced (Tako-Tsubo) cardiomyopathy [21].
Acute left ventricular (LV) failure is a rare manifestation of acute cardiotoxicity, but it could be reversible with appropriate prevention.
Generally arrhythmias associated with acute anthracycline administration are transient and do not require specific interventions [57]. However it has been reported that patients with syncope and complete heart block require pacemaker implantation during anthracycline therapy at only modest doses (cumulative dose of 120 mg/m2) [66]. Also for epirubicin, which seems to have a more favorable cardiovascular toxicity profile, this kind of adverse effect has been described.
It is still unclear if the patients that experienced these early side effects are more prone than the patients without these to the occurrence of late toxicity.
Heart failure is usually a late event, generally extending the risk over a period of years.
As previously shown the incidence of chronic doxorubicin cardiotoxicity is much lower (about 1.7 %) [22] and related to the dose.
Early-onset chronic progressive cardiotoxicity appears after 1 week and within 1 year after completion of therapy, and it is usually evident within 30 days of administration of its last dose, with a probability of 1.6–2.1 %. A late-onset chronic cardiotoxicity may occur after the first year, and it can develop even after 6–20 years after chemotherapy, and it is unclear whether there is any time limit for its development.
Changes in left ventricular ejection fraction (LVEF) with anthracycline chemotherapy are evident at cumulative doses of doxorubicin >350 mg/m2. Generally these decreases in left ventricular function are asymptomatic, and if the cardiotoxicity is no more than moderate (a decrease in LVEF of ≥15 % to an absolute LVEF between 30 and 45 %), stabilization of the LVEF can occur with discontinuation of anthracycline therapy.
Cardiotoxicity increases in long-term survivors, from 2 % after 2 years to 5 % after 15 years.
This particular aspect is relevant in adult survivors of pediatric malignancies. Up to 65 % of patients with a history of childhood malignancy treated with doxorubicin can have echocardiographic evidence of left ventricular contractile abnormalities [23].
In the Childhood Cancer Survivor Study, a study of 14,358 5-year survivors of childhood malignancies, use of <250 mg/m2 of anthracycline was associated with a 2.4-fold higher risk of developing congestive heart failure compared to those patients who did not receive anthracyclines [24]. This risk increased to 5.2-fold with the use of ≥250 mg/m2 of doxorubicin.
In survivors of childhood Hodgkin’s lymphoma analyzed by Doppler echocardiography [67] was found a high prevalence of diastolic dysfunction, evaluating different echocardiography variables, even in asymptomatic survivors, confirming that diastolic dysfunction is common after anthracycline therapy, even in patients not submitted to radiotherapy and even with a median dose of doxorubicin of 150 mg/m2 with 69 % of patients receiving <300 mg/m2, cutoff commonly retained adequate to designate the highest risk for systolic dysfunction [68].
In adult patients with breast cancer treated with adjuvant chemotherapy that included anthracyclines, Abu-Khalaf et al. demonstrated that the median absolute change in LVEF from baseline was −5.5 % 7 years after receiving anthracyclines [24]. Furthermore, 12 % of their cohort had an LVEF below the lower limit of normal following chemotherapy.
A relationship between ventricular systolic dysfunction and arrhythmogenesis in anthracycline cardiotoxicity as supposed by an investigation including 72 adults treated with anthracycline has been hypothesized [69]. A moderately good correlation between the QTc interval during anthracycline therapy and the degree of LV enlargement (r = 0.43) and with ejection fraction (r = −0.46), with a negative correlation indicating the association of the reduction in ejection fraction with longer QTc intervals, was found. There were no associations between QTc interval and LV diastolic function, commonly considered the first step in myocardial damage after anthracycline administration. However in 1981 a case report of sudden cardiac death of a patient after the administration of a high cumulative dose of doxorubicin (490 mg/m2) 8 months before without evidence of CHF or history of arrhythmia was described [70]. Cardiac fibrosis and hypertrophy were shown at postmortem analysis, suggesting that the lack of overt clinical heart failure could not exclude marked myocardial dysfunction at the time of the arrhythmic death.
The prognosis of patients who develop congestive heart failure is poor with approximately 50 % of mortality in the first year.
2.3.1.6 Diagnosis of Anthracycline Toxicity
The first step in the diagnosis of doxorubicin cardiomyopathy should consist in taking an appropriate history to assess the likelihood of the diagnosis.
The complete examination of the cardiovascular system is essential to detect the presence of signs of overt heart failure, such as elevated jugular venous pressure and S3 gallop.
In every patient the electrocardiogram should be obtained: usually the ECG demonstrates nonspecific ST–T wave changes and sometimes low-voltage QRS complexes, less commonly the alterations previously described in cardiac rhythm and in ventricular repolarization.
A chest X-ray may also be helpful to assess cardiomegaly and signs of pulmonary venous congestion.
The abnormalities shown by means of these evaluations are nonspecific and nondiagnostic. Antimyosin antibody search with the use of 111In-labeled monoclonal antimyosin antibody is used for the diagnosis of myocarditis and has also been employed for the diagnosis of doxorubicin cardiomyopathy. In doxorubicin-treated patients, the sensitivity of antimyosin antibody studies is very high [49].
Various histology nuclear studies have been attempted to detect apoptosis in animal models. Annexin V has been used to detect apoptosis induced by doxorubicin.
With greater extent the measurement of neurohormones and cardiac enzymes for the diagnosis of doxorubicin cardiotoxicity and presence of heart failure has been studied.
Plasma levels of B-type natriuretic peptide have been found elevated correlating with the severity of congestive heart failure.
Also the elevation of troponin T or I levels has been shown in different studies, indicating myocardial damage.
The changes in neurohormones and cardiac enzymes however are observed in other types of cardiomyopathy and are not specific of doxorubicin cardiomyopathy.
Endomyocardial biopsy may reveal the characteristic diagnostic features of doxorubicin cardiomyopathy described in the paragraph on cardiac morphology and histopathological modifications. The findings suggested for the diagnosis of doxorubicin cardiomyopathy are loss of myofibrils, distention of sarcoplasmic reticulum, and vacuolization of the cytoplasm (the so-called Adria cells). The endomyocardial biopsy may also be used to evaluate the severity of doxorubicin cardiotoxicity. However this technique is invasive, and it requires considerable experience and training; as a consequence, endomyocardial biopsies are not universally used for the diagnosis of doxorubicin cardiomyopathy and its severity.
2.3.1.7 Assessment of Left Ventricular Function
Left ventricular systolic and diastolic function can be assessed with a variety of noninvasive imaging techniques, including transthoracic echocardiography (ECHO), equilibrium radionuclide angiography (RNA), cardiac magnetic resonance (CMR), and cardiac computed tomography (CT). For the specific use of these imaging techniques, see the Chap. 4.
The methods commonly used in clinical practice to monitor left ventricular function in patients receiving potentially cardiotoxic chemotherapy are ECHO and RNA.
RNA is count based and therefore quantitative, but the patient is exposed to a radiation of approximately 5 millisieverts also if newer gamma cameras employing more sensitive crystals may lead to dose reductions, with a radiation exposure fivefold inferior. Radionuclide ventriculography has been used to assess both LV systolic and diastolic functions.
Like in other kinds of cardiomyopathies, cardiac adrenergic denervation can occur in doxorubicin cardiomyopathy, and the use of MIBG (metaiodobenzylguanidine) nuclear imaging can be employed to assess this cardiac adrenergic denervation.
Doxorubicin cardiomyopathy has shown impairment in glucose and fatty acid metabolism. Impaired myocardial glucose uptake can be assessed by positron emission tomography (PET) using fluorine-18F-deoxyglucose. Abnormal fatty acid metabolism can be evaluated by 123I-BMIPP (β-methyl-branched fatty acid) nuclear studies.
ECHO imaging, in contrast, does not require radiation exposure, and an accurate quantitative measure of LVEF can be obtained in all patients with a good echographic window if careful planimetry of the left ventricle is performed. Moreover echocardiography with Doppler studies is commonly used to detect early diastolic and systolic LV dysfunction.
Exercise echocardiography or pharmacologic ECHO stress may also be useful to assess LV contractile reserve.
In addition, echocardiography provides additional anatomic assessment of the myocardium, valve structure, and great vessels that cannot be obtained by RNA.
In patients without a good echo window who cannot show good images, use of echo contrast material or RNA should be used to assess LVEF.
While either ECHO or RNA may be chosen to assess left ventricular function in patients receiving chemotherapy, it is essential during follow-up in serial examinations the use of the same technique to avoid variability between the two methods.
Also cardiac magnetic resonance imaging can be used to assess LV systolic function. These tests however are not specific for doxorubicin cardiomyopathy.
For more information see Chap. 4.
The reassessment of cardiac function is important in all patients after treatment with anthracyclines to identify asymptomatic patients who are undergoing progressive cardiac damage; if it has been shown that an LVEF decreases by either 15 percentage points or 10 percentage points to a value below 50 and a repeat assessment after 3 weeks confirms these findings, or if a troponin or BNP elevation has been shown, alternative chemotherapeutic options have to be considered, as continuing treatment with an anthracycline entitles an unacceptable risk for cardiotoxicity.
There is at present no clear consensus regarding the duration of the follow-up for asymptomatic patients. A measurement of systolic function at 6 months after the conclusion of treatment, annually for 2 or 3 years thereafter, and then at 3- to 5-year intervals for life could be reasonable. If during the follow-up a CV event should occur, the controls should be more stringent. High-risk patients, e.g., those with underlying CV disease or those who have received >300 mg/m2 of doxorubicin or equivalent, should be monitored more frequently, although until now no data have been reported supporting an outcome advantage resulting from this strategy.
Box 2.3: Anthracycline Toxicity Clinic
Dose relationship for cardiac toxicity
Specific risk factors
Cardiomyopathy with specific findings (Adria cells)
Toxicity: imbalance between increased oxidative stress (mitochondrial failure – SERCA dysfunction – CA overload – caspase activation)/cytoprotective pathways (erbB2 and Akt activation)
Acute toxicity immediately after administration (incidence 11 %)
Chest pain, SVPB, VPB, SVT, and nonspecific ST/T changes
Late toxicity from 30 days to 10 years (incidence 1.7 %)
Diagnosis based on history, symptoms, and findings of heart failure – increase of neurohormones and troponin (not specific)
Assessment of LVEF by ECHO and radionuclide angiography
2.3.1.8 Prevention of Anthracycline Cardiac Toxicity [30–32, 50]
According to the ACC and AHA guidelines on heart failure, patients receiving chemotherapy may be considered a Stage A HF group, with an increased risk of developing cardiac dysfunction. Βeta-blockers and ACE inhibitors are the drugs of choice in this patient category.
Considering that main anthracycline-induced cardiotoxicity mechanisms are probably due to generation of free radicals in the cardiomyocytes resulting in apoptosis and consequent left ventricular dysfunction and heart failure, efforts aimed at decreasing this exogenous oxidant stress may decrease the cardiotoxicity of anthracyclines and attenuate the left ventricular dysfunction associated with the use of this agent. This goal can be achieved through a variety of mechanisms. Reactive oxygen species can be generated by the interaction of doxorubicin with nonheme iron. Among beta-blockers carvedilol may prevent cardiac damage induced by doxorubicin also by means of its antioxidant activity. In patients receiving high doses of doxorubicin (>500 mg/m2), treatment with carvedilol prevented the decline in LVEF. The effect of carvedilol has been confirmed in a randomized study in which prophylactic use of carvedilol in a population of patients treated with anthracycline prevented left ventricular dysfunction and reduced mortality [71]. Valsartan, an angiotensin receptor blocker (ARB), given concurrently to anthracycline-containing regimens, has been shown to prevent acute cardiac damage [72].
In one study ranolazine prevented echocardiographic and/or biohumoral indices of diastolic dysfunction, measured at 5 weeks or 6 months after the administration [73].
Other agents, particularly in patients at increased risk, have shown promising results in reducing cardiotoxicity. Patients with increased plasma troponin I during treatment with doxorubicin are a risk patient category. The treatment with enalapril prevented the late decline in LVEF in these patients.
Therefore treatments with ACE inhibitors and/or carvedilol have to be considered in patients at risk for the development of anthracycline cardiotoxicity.
Also the antioxidant probucol has been shown to prevent the decrease in LVEF in animal models of doxorubicin cardiotoxicity.
Otherwise the antioxidant properties of vitamin E have not been shown to be effective in preventing left ventricular dysfunction in animals treated with doxorubicin.
The iron-chelating agent dexrazoxane has shown the possibility to reduce anthracycline-induced cardiac apoptosis by the inhibition of iron-catalyzed radical generation by means of the superoxide dismutase mimetic properties both in adult and in pediatric populations. Due to inhibition of reactive oxygen species generation, this iron-chelating agent was introduced as a preventive treatment of anthracycline-induced cardiotoxicity. This agent has been shown in multiple trials to reduce the incidence of congestive heart failure and decreases in LVEF. The cardioprotective effect of dexrazoxane has been observed even after patients have received 300 mg/m2 of doxorubicin. This agent seems to reduce the risk of anthracycline cardiotoxicity by about two-thirds, without affecting response to chemotherapy or overall survival, and has shown in children the ability to prevent the elevation of troponin T secondary to the treatment.
The use of this agent remains limited by the doubts suggested by some studies hypothesizing a reduction of the oncologic therapeutic effect of the anthracycline by its concomitant administration. These concerns led the US Food and Drug Administration to recommend the use of dexrazoxane only at high dosages of anthracyclines (higher than 300 mg/m2 for doxorubicin). Other investigations have not confirmed a reduction in oncologic efficacy by its use.
The vinca alkaloid vincristine exerts cardioprotective effects on cultured adult mouse cardiac myocytes to chemical and hypoxic oxidative stress and is often used in combination with doxorubicin to enhance the effectiveness for treatment of the malignancy [51]. Exposure of cultured myocytes to 15 or 20 μg/ml of doxorubicin for approximately 24 h typically reduces myocyte survival by 50 %. Co-treatment with 10–30 μmol/l of vincristine dramatically increased cardiomyocyte survival (>85 %). Compared to doxorubicin treatment alone, co-treatment with vincristine decreased cytochrome C release, suggesting that vincristine decreases oxidative stress and inhibits mitochondrial transition. The effects of vincristine were compared to those of mercaptopropionyl glycine (which has antioxidant properties), amlodipine (a dihydropyridine calcium channel blocker), and dexrazoxane (an iron-chelating agent): they were superior to mercaptopropionyl glycine and amlodipine and similar to dexrazoxane.
During that time other several methods have been evaluated to reduce the anthracycline toxicity. Modification of pharmacokinetic characteristics by liposomal encapsulation showed that daunorubicin and doxorubicin are less toxic to cardiac tissue than the non-liposomal form because a lower proportion of drug is administered in the liposome form to the heart. Delivery of doxorubicin in a pegylated liposomal form decreases the circulating concentrations of free doxorubicin and results in selective uptake of the agent in tumor cells [29]. Use of pegylated liposomal doxorubicin has been shown to decrease the cardiotoxicity of this anthracycline, even at doses >500 mg/m2.
Another possibility to reduce toxicity has been considered in the modification of the chemical structure (like for epirubicin) as described before.
Finally modifications of drug infusion regimens studying different protocols to allow a reduction in peak plasma levels of drugs using longer infusion rates have been proposed.
2.3.1.9 Prevention Strategy of Anthracycline Cardiac Toxicity
The decision to treat a malignancy using a chemotherapy enclosing an anthracycline must evaluate the ratio between the potential benefit of the treatment and the potential cardiac risk. This judgment requires a deep discussion between the cardiologist and the oncologist to establish pros and contras of anthracycline therapy. If the patient has no or reduced risk factors for anthracycline-induced cardiotoxicity and if he/she will benefit greatly from treatment with anthracyclines, the decision to utilize this drug family is positive. A routine monitoring of left ventricular function is required. On the other end patients with high risk for anthracycline-induced cardiotoxicity and patients with an uncertain benefit from the use of anthracycline should not receive these agents. In the intermediate category of patients, the relative risk and benefits of anthracycline use have to be carefully evaluated and will determine the decisions whether to treat or not to treat submitting these subjects to more frequent monitoring (e.g., before every cycle of chemotherapy) and to the preventive treatment of cardioprotective agents.
Similar evaluations of the risk and benefit must be made in patients that develop anthracycline-induced cardiotoxicity during treatment, and it must be decided whether to continue or interrupt the therapy. In this scenario, perhaps the degree of left ventricular dysfunction can play a significant role in determining whether to continue treating with an anthracycline agent. In patients who may benefit greatly from a chemotherapy regimen that includes doxorubicin, it may be decided to continue the therapy if the LVEF is >40 %, with the association of cardioprotective treatments and performing frequent monitoring of left ventricular function.
The major concentration has been the limitation of the cumulative dose of doxorubicin to <450 mg/m2. As previously said the use of anthracycline analogues, alternative methods of drug delivery, and continuous slow infusion instead of standard infusion protocols are also methods employed to reduce the risk of dilated cardiomyopathy. However at present, clear universally accepted strategies to prevent or reduce doxorubicin cardiotoxicity have not been established.
Box 2.4: Pharmacologic Agents Used to Reduce Doxorubicin Cardiotoxicity
MPG
Probucol
Dexrazoxane
Amlodipine
Carvedilol
PDE5 inhibitor (sildenafil)
Nitric oxide
Superoxide dismutase
Endothelin receptor antagonist (bosentan)
Erythropoietin and thrombopoietin
Granulocyte colony-stimulating factor
Vincristine
Many of these agents have been tested in experimental animal studies to evaluate a possible role in reducing the risk of doxorubicin cardiotoxicity. Most of these agents have an effect in reducing oxidative stress. Mercaptopropionyl glycine (MPG), a synthetic aminothiol that exhibits antioxidant properties, probucol, superoxide dismutase, and dexrazoxane with documented antioxidant properties have been reported to decrease doxorubicin cardiotoxicity. Also amlodipine and the β- and α-adrenergic blocking agent carvedilol that have antioxidant properties have been studied. The PDE5 inhibitor sildenafil, erythropoietin and thrombopoietin, granulocyte colony-stimulating factor, and the endothelin receptor-blocking agent bosentan have been analyzed in experimental animal models. The potential protective role of nitric oxide and superoxide dismutase has been investigated in a transgenic mouse model. Lack of nitric oxide was associated with enhanced cardiac injury, and mitochondrial injury was attenuated by an increase in manganese superoxide dismutase.
However the potential protective effect against doxorubicin cardiotoxicity has been studied in animals, usually during acute and short-term exposure to doxorubicin employing a short-term intraperitoneal administration. Various methods to assess cardiotoxicity have been used in different studies. The protective effects of vincristine may differ according to the species, and it remains unknown whether vincristine will exert similar effects in humans. Appropriate clinical trial needs to be used to demonstrate whether any of these agents will be useful in the treatment of doxorubicin cardiomyopathy. Although the iron-chelating agent dexrazoxane is available for clinical use, in practice it is used infrequently because it can induce myelosuppression.
In conclusion, doxorubicin cardiomyopathy remains a lethal disease. Extensive investigations and research have been done to understand the mechanism of doxorubicin cardiotoxicity, and substantial progresses in its knowledge have been reached. Although extensive research has also been expanded to find effective treatment of doxorubicin cardiomyopathy, a definite treatment has been not yet discovered.
2.3.1.10 Treatment and Management
When anthracycline-induced heart failure has been established, there are no specific treatments available for the management of these patients. Standard therapies for congestive heart failure according to the current guidelines of ACC and AHA and of ESC should utilize ACE inhibitors, beta-blockers, and loop diuretics for volume management.
Diuretics are used to relieve symptoms and signs of pulmonary and systemic venous congestion. Β-Adrenergic blocking agents should be considered, as for treatment of other types of systolic heart failure. Apart from the previously discussed carvedilol, it has been reported that metoprolol is safe and can be effective in doxorubicin-induced cardiomyopathy. However there are few controlled data whether β-blocker treatments are effective to prevent progression of remodeling and to improve prognosis, and there are also few information about the change in prognosis of doxorubicin cardiomyopathy before and after introduction of β-blocker therapy.
Angiotensin II inhibition should also be considered. Recently treatment with enalapril and carvedilol in a study of patients with an anthracycline-induced decrease in LVEF ≤45 % resulted in normalization of LVEF in 42 % of patients. These responders had a higher LVEF after the onset of congestive heart failure compared to partial responders (LVEF increased >10 % but did not normalize) and nonresponders and had therapy initiated sooner than partial responders and nonresponders [52]. In population of anthracycline-induced cardiomyopathy patients, it has been demonstrated that an essential variable for the recovery of cardiac dysfunction is the time from the end of chemotherapy to the beginning of HF therapy (time to treatment) with ACE-I and, when tolerated, with BB. Likelihood of obtaining a complete LVEF recovery is higher in patients initiating treatment within 2 months from the end of chemotherapy.
In patients with advanced heart failure and in those intolerant to angiotensin II inhibition therapy, low-dose hydralazine–isosorbide dinitrate combination treatment is possible; however, there is no information available to suggest that such treatment is effective in patients with doxorubicin cardiomyopathy.
In patients with malignant arrhythmias, amiodarone and implantable cardioverter and defibrillator, in patients without a prognosis of mortality in short time as stated in the relative guidelines, should be considered.
However it has to be underlined that none of the treatments employed for ischemic or idiopathic dilated cardiomyopathy has been demonstrated to improve the prognosis of patients with doxorubicin cardiomyopathy.
Finally cardiac transplantation has been reported to improve long-term prognosis of the patients in whom the primary malignancy is cured following chemotherapy, and the implantation of ventricular assist devices may be required as bridge strategy before cardiac transplantation. The same limitations to transplant and assist devices are applied as for AICDs.
In patients who develop left ventricular dysfunction during or after receiving anthracycline-based chemotherapy, it is important to consider other causes of heart failure. In particular, coronary artery disease must be considered in adult patients with risk factors for coronary artery disease and an ischemia workup initiated. In addition, endomyocardial biopsy may be considered in patients in whom there is question as to the cause of left ventricular dysfunction or in helping to determine if anthracycline chemotherapy should be continued, especially if the patient has received high doses of the agent.
Unfortunately it is particularly important to observe that, as for many other fields of preventive medicine, only about one-third of patients receiving chemotherapy with an asymptomatic decrease in LVEF receive an ACE inhibitor or angiotensin receptor blocker and/or a beta-blocker and less than a half are considered for cardiology consultation. This last fact once again emphasizes the importance of communication between the oncologist and cardiologist and a common education regarding the problem of cardiac toxicity after chemotherapy.
Cardiac monitoring in patients with overt LVEF impairment is recommended at 3, 6, and 9 months.
Box 2.5: Prevention and Management of Anthracycline Toxicity
Evaluate ratio potential benefit of the treatment/potential cardiac risk
Limit the cumulative dose of doxorubicin to <450 mg/m2
Antioxidant agents
Beta-blockers and ACE inhibitors in HF and in asymptomatic decrease of LVEF
Cardiac monitoring at 3, 6, and 9 months
2.3.1.11 Mitoxantrone
Mitoxantrone is an anthracenedione antineoplastic agent with a chemical structure similar to other anthracyclines.
Cardiotoxicity is a severe side effect also of mitoxantrone.
The risk of mitoxantrone-induced cardiotoxicity has been studied also in patients with multiple sclerosis, and it increases with cumulative doses >100 mg/m2 body surface area (BSA). However, the effect of mitoxantrone on cardiac function during treatment with cumulative doses <100 mg/m2 BSA, mainly in the early phases, is unclear, and few data on cardiac function regularly monitored from the beginning of treatment are available.
Doxorubicin, which has an anthracycline structure, alters Ca2+-releasing and uptake mechanisms in the sarcoplasmic reticulum of myocardial cells, and, as previously described, these effects are apparently one of the possible effects related to anthracycline cardiotoxicity.
Mitoxantrone is however significantly less cardiotoxic. The effects of mitoxantrone examined in preparations of the electrically stimulated left atrial muscle on the functions of the sarcoplasmic reticulum preparations suggest that mitoxantrone increases Ca2+ release. Apparent enhancement of the sarcoplasmic reticulum functions, in contrast to anthracyclines that has been shown to suppress these functions, seems to explain its relative less marked cardiotoxicity.
The therapeutic benefit of mitoxantrone for patients with worsening multiple sclerosis has been proved in clinical trials. A retrospective analysis of 1,378 patients with multiple sclerosis showed an asymptomatic decrease of LVEF to <50 % in 1.8 % of patients below a cumulative dose of 100 mg/m2, compared with 5 % above 100 mg/m2, without continuous monitoring of cardiac function from the beginning of treatment. By performing regular echocardiography before each infusion, 22 % patients transiently experienced a notable decrease in LVEF of between 13 and 16 % after their first or second mitoxantrone infusion. Another 22 % of patients showed diastolic changes without marked changes in LVEF [74].
During mitoxantrone administration also some episodic case of an acute myocarditis-like clinical picture and also the occurrence of arrhythmias has been shown.
Box 2.6 a: Anthracyclines (1)
Still among the most effective and used chemotherapy agents
Dose relationship with toxic effect (max 240–360 mg/m2 of doxorubicin and 450–600 mg/m2 of epirubicin)
Association with trastuzumab toxic effect
Risk factors for toxicity (see Table 2.1)
Genetic factors predisposing to toxicity
Toxic mechanisms:
Increased oxidative cell stress and ROS production
Mitochondrial dysfunction alterations in calcium homeostasis and contraction
Alteration of the sarcoplasmic reticulum, protein contractile degradation, and sarcomere disruption
Cell apoptosis activation
Imbalance between apoptotic and protective pathways
Box 2.6 b: Anthracyclines (2)
Acute cardiotoxicity 11 % during administration or 2–3 days
Myopericarditis, chest pain, PVCs, PSVCs, NS-SVT, ST changes, Tako-Tsubo cardiomyopathy
Chronic cardiotoxicity 1.7 % not clearly related to acute toxicity, within 30 days to 6–10 years from administration
Special population – survivors to childhood malignancies
Poor prognosis (50 % mortality at 1 year)
Diagnosis
Assessment of LV function
Prevention
Treatment
2.3.2 Fluoropyrimidines (5-FU and Capecitabine)
5-Fluoropyrimidines and capecitabine are antimetabolites inhibiting thymidylate synthase and, as a consequence, the production of thymidine and uracil. The unavailability of this basis inhibits the synthesis of RNA and DNA and stops the growth and division of rapidly dividing cells like tumor cells.
Cardiac toxicity has been estimated from 1.2 to 18 % of treated patients, often misdiagnosed, with a mortality of 8 % and a further increase of 13 % in reexposed subjects.
It can occur with a wide dose range.
Predisposing factors are a preexisting coronary heart disease, the genetic deficiency of dihydropyrimidine dehydrogenase (present in 8 % of normal population) that prevents the possibility to metabolize 5-FU, and the supplementation of folic acid in diet that increases the levels of methylenetetrahydrofolate which stabilizes 5-FU to thymidylate synthase binding.
The histologic damages of 5-fluoropyrimidines are under debate and are comprehensive of the classic ischemic damages but also cytotoxic direct damages.
The mechanism of toxicity is related to the induction of endothelial damage with a reduction of NO synthase leading to vasospasm and to cytotoxic damage with leakage of the endothelium with possible induction of thrombosis. This mechanism is fully reversible.
Clinical expressions of these lesions are typical angina, myocardial infarction, and arrhythmias, generally from the first hour to 48 h after the administration of the drug; these symptoms disappear after discontinuation of the therapy.
Electrocardiographic monitoring is essential both for the prevention and for the immediate diagnosis of toxicity. Once this has been arisen, the immediate suspension and replacement with a different chemotherapy class agent are mandatory.
During symptoms the treatment consists of verapamil and nitrates active against coronary spasm.
2.3.2.1 Introduction
5-Fluorouracil or 5-FU belongs to the family of chemotherapeutic drugs called antimetabolites, and it is a pyrimidine analogue acting through irreversible inhibition of thymidylate synthase. 5-FU blocks the synthesis of the thymidine, a nucleoside required for DNA replication, interrupting the action of this enzyme and inhibiting the transformation of deoxyuridine monophosphate (dUMP) into thymidine monophosphate (dTMP). The shortage of dTMP causes the death of the rapidly dividing cancerous cells. Since uracil is a normal component of RNA, the rationale in developing the drug was that cancer cells, with their increased genetic instability, might be more sensitive to toxic molecules that mimic the natural compound than normal cells. Synthesizing a drug with a specific uracil antagonism was the scientific challenge in this perspective. 5-Fluorouracil is obtained by reacting elemental fluorine with uracil. 5-FU was produced and patented in 1957 and has been used against cancer until now.
The drug substitutes uracil during the nucleic acid replication process. Because 5-fluorouracil is similar in shape to but does not maintain the same chemical characteristics as uracil, the drug inhibits RNA replication enzymes, thereby blocking RNA synthesis and stopping the growth of cancerous cells. As a pyrimidine analogue, it is transformed by the cell into different cytotoxic metabolites which are incorporated into nucleic acids, provoking the interruption of the cell cycle and apoptosis inhibiting DNA synthesis. The drug is acting only during the S phase of the cell cycle. Furthermore it has been shown that 5-FU inhibits the activity of the exoribonuclease contained in the exosome complex that is essential for cell survival.
2.3.2.2 Epidemiology of Fluoropyrimidine Toxicity
The antimetabolite fluorouracil (5-FU) is frequently administered for chemotherapy of various malignant neoplasms. Capecitabine is a prodrug administered orally that converts preferentially within tumors to 5-FU, resulting in enhanced concentrations of 5-FU in the target neoplastic tissues. Through this specific action, capecitabine was expected to reduce the risk of side effects associated with fluoropyrimidine.
5-Fluorouracil (5-FU) and its oral prodrug capecitabine are mainly used in oncology for the treatment of various solid tumors, including colorectal cancers.
Well-known adverse effects of the drug involve the bone marrow, skin, mucous membranes, intestinal tract, and central nervous system, whereas cardiac toxicity of 5-FU is less familiar to clinicians. Its cardiac side effects have an incidence variable in the literature from 1.2 to 7.6 %. In other series cardiotoxicity due to 5-FU and capecitabine is not an uncommon adverse effect and has been reported with a maximum incidence of 18 % of patients.
Potentially, arrhythmias, myocardial infarction, and sudden cardiac death could occur.
There are conflicting data on overall mortality due to cardiac toxicity of fluoropyrimidines: from a life-threatening cardiotoxicity observed with a frequency <1 % [53] to an overall mortality rate estimated to be between 2.2 and 13.3 % [54].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


