Characteristics
RV
LV
Shape (cross section)
Crescentic
Elliptic
End-diastolic volume (ml/m2)
49–101
44–89
Ventricular pressures
(range mmHg)
(15–30)/(1–7)
(90–140)/(5–12)
RVEF (%)
>40–45 %
>50 %
Ventricular elastance (E max) mmHg/ml
1.3 ± 0.84
5.48 ± 1.23
PVR vs. SVR, dyne.s.cm−5
20–130
700–1,600
Stroke work index, g/m2 per beat
8 ± 2
50 ± 20
As is the case for the LV, the major determinants of RV systolic function are myocardial contractility, preload, and afterload. The RV effective output is influenced by heart rhythm, intraventricular synchrony, and ventricular interdependence [14]. Ventricular interdependence refers to the concept that the size, shape, and compliance of one ventricle may affect the performance of the other ventricle. It is recognized an essential part in the pathophysiology of RV dysfunction [15]. It is most apparent with changes in loading conditions, such as those seen with respiration or sudden postural changes. Three factors determine ventricular interdependence: (1) the interventricular septum, (2) the pericardium, and (3) muscular fiber continuity between the LV and RV. Systolic ventricular interdependence is mediated mainly through the interventricular septum, while the pericardium contributes mainly to diastolic interdependence [16]. Bernheim, almost 100 years ago, was the first who hypothesized the ventricular interdependence with the concept that alterations of one ventricle could affect the other through the myocardium [17]. Half a century after, Dexter described deterioration of LV function in patients with atrial septal defects (ASDs) who developed RV pressure and volume overload. He called this the “reverse Bernheim effect,” postulating leftward septal shift with resultant impaired LV filling and function [18].
It has been estimated in experimental studies that 20–40 % of RV systolic pressure and volume outflow results from LV contraction [14]. In situations where a part of the RV is akinetic, due to scarring post myocardial infarction or after replacement with a noncontractile material (i.e., synthetic patch), the septum is able to maintain circulatory stability as long as the RV is not dilated [19]. In acute RV pressure- or volume-overload states, dilatation of the RV increases intrapericardial pressure and shifts the interventricular septum to the left, altering LV geometry. As a consequence, distensibility of LV decreases, leading to a decreased LV preload, increased LV end-diastolic pressure, and consequently a low cardiac output state (Fig. 3.1) [4]. Conversely, LV volume or pressure overload has also been shown to redistribute RV filling into late diastole. Diastolic ventricular interdependence occurs even with the pericardium opens, as after cardiac surgery, although the coupling is augmented with it closed [20].
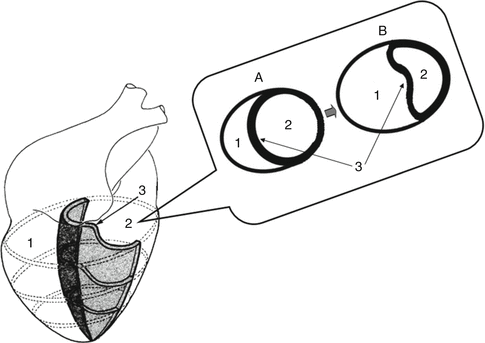
Fig. 3.1
Ventricular interdependence in RV failure. Cross section (A) represents normal anatomy. Note the shift of the interventricular septum toward the left (B), changing LV geometry and ultimately reducing cardiac output. (1) Right ventricle, (2) left ventricle, (3) interventricular septum
The complex relationship between RV contractility, preload, and afterload can be better understood with the help of pressure–volume loops. Pressure–volume loops depict instantaneous pressure–volume curves under different loading conditions (Fig. 3.2). The slope of the end-systolic pressure–volume relationship is referred to as ventricular elastance (E max). Because of its relative load independence, many investigators consider ventricular elastance as the most reliable index of contractility [22]. On the basis of the studies by Dell’Italia and Walsh, the normal maximal RV elastance is 1.3 ± 0.84 mmHg/ml, while for the LV is four times higher [23, 24]. Consequently, the RV is far more sensitive to increases in afterload. This can be illustrated in the acute setting, where RV stroke volume decreases significantly after an increase in pulmonary arterial pressure.
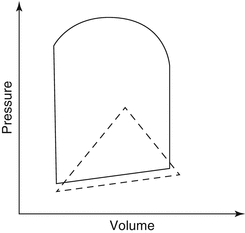
Fig. 3.2
Pressure–volume loops for the RV and LV. The pulmonary valve opens early in systole once RV pressure reaches the (relatively low) pulmonary pressure. Little time is spent in isovolumic contraction, giving a triangular-shaped pressure–volume loop, in contrast to the almost square loop of the LV (Adapted from Kevin and Barnard [21])
RV preload represents the load present before contraction. Under normal conditions, an increase in RV preload improves myocardial contraction on the basis of the Frank–Starling law, while in conditions of RV failure, this curve flattens and moves downward and to the right (Fig. 3.3). As previously analyzed, excessive RV volume loading compresses LV filling and impairs global ventricular function through the mechanism of ventricular interdependence (Fig. 3.1). Factors that influence RV filling include intravascular volume status, ventricular compliance, heart rate, LV filling, and pericardial constraint [25]. The pericardium imposes greater constraint on the thinner, more compliant, low-pressure RV. The filling period is also an important determinant of RV preload. In post-extrasystolic beats, the filling period is longer, leading to increased stroke volume. Factors that affect right ventricular preload, afterload, or left ventricular function can adversely influence the functioning of the RV, causing ischemia and RV failure. As RV failure progresses, a pronounced tricuspid regurgitation further decreases cardiac output and worsens organ congestion. This can degenerate into an irreversible vicious cycle [26].
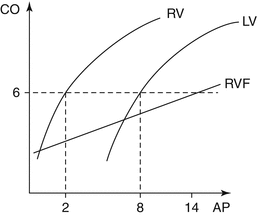
Fig. 3.3
Comparison of RV and LV Starling curves. LV requires higher atrial filling pressures (AP) to produce equivalent cardiac output (CO). In RV failure (RVF) the curve moves downward and to the right
Afterload sensitivity represents a major physiologic characteristic of RV as compared to LV (Fig. 3.4). This is attributed primarily to its thinner muscular wall. The major determinant of RV afterload is pulmonary vascular resistance (PVR). The pulmonary vascular bed represents a highly compliant, low-pressure, low-resistance system. In the presence of normal pulmonary circulation, the RV performs approximately one-fourth of the LV stroke work [28]. Several factors modulate PVR, including hypoxia, hypercarbia, cardiac output, and pulmonary volume and pressure. Pulmonary vessels constrict with hypoxia, known as Euler–Liljestrand reflex, and hypercarbia and relax with hyperoxia [29]. Lung volumes exert a significant effect on PVR. As illustrated in Fig. 3.5, the relationship between lung volume and PVR has a unique U-shape. Thus, PVR is minimal at functional residual capacity and increases both at large and at small lung volumes due to compression of alveolar and extra-alveolar vessels. For example, in patients with pulmonary emphysema as well as with acute respiratory distress (ARDS), hyperinflation of the lungs greatly increases PVR [29].
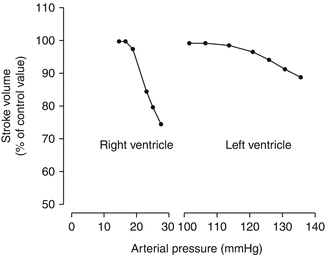
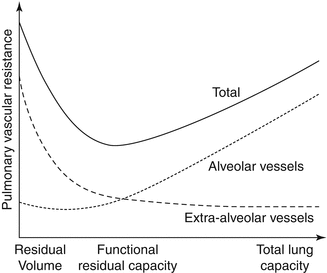
Fig. 3.4
The response of the right and left ventricles to experimental increase in pressure or afterload (Adapted from MacNee [27])
Fig. 3.5
Relationship between lung volumes and pulmonary vascular resistance (PVR). PVR is minimum at functional residual capacity and increases at large and small volumes (Adapted from Fischer et al. [29])
3.1.3 Heart Rhythm and RV Function
The RV is particularly sensitive to changes in heart rhythm and synchrony with the LV. In the presence of RV dysfunction, maintenance of sinus rhythm and AV synchrony is crucial for producing an effective cardiac output. Atrial fibrillation or complete AV block are poorly tolerated in the setting of an acute RV myocardial infarction or acute pulmonary emboli [30]. RV dyssynchrony refers to the concept of suboptimal coordination of RV mechanical function in relation to LV. An increase in RV wall tension with time is associated with a longer duration of RV ejection which causes RV contraction to continue beyond LV contraction and leads to RV systolic phase upon LV filling [31]. RV dyssynchrony could potentially lead to reduced cardiac output or increased filling pressures. Thus, resynchronization therapy is associated with an increase in RV ejection fraction in patients with RV failure [32].
3.2 Pathophysiology of Right Heart Failure
The RV may be subject to pressure or volume overload, ischemia, intrinsic myocardial disease, or pericardial constraint. Conditions leading to right heart failure related to the underlying mechanism are described in Table 3.2. According to the pathophysiology leading to RV failure, different etiologies can be grouped into three main categories: (1) RV failure secondary to increased RV afterload, (2) RV failure because of volume overload, and (3) intrinsic RV failure in the absence of pulmonary hypertension (usually RV infarction).
Table 3.2
Mechanisms and specific causes of right heart failure
Pressure overload |
Left-sided heart failure |
Pulmonary embolism |
Pulmonary arterial hypertension |
Pulmonary stenosis |
RV outflow tract obstruction |
Volume overload |
Tricuspid regurgitation |
Pulmonary regurgitation |
Atrial septal defect |
Anomalous pulmonary venous return |
Carcinoid syndrome |
Rheumatic valvular disease |
Ischemia and infarction |
RV myocardial infarction |
Complex congenital defects |
Ebstein’s anomaly |
Tetralogy of Fallot |
Transposition of great arteries |
Systemic RV |
Double-outlet RV |
Intrinsic myocardial disease |
Cardiomyopathies |
Arrhythmogenic RV dysplasia |
Sepsis |
Inflow limitation |
Tricuspid stenosis |
Superior vena cava stenosis |
Pericardial disease Constrictive pericarditis |
3.2.1 The RV Under Pressure: Adaptive Hypertrophy
The RV is thinner than the LV and has a different shape, which reflects the low pressure in the pulmonary circulation and allows quick adaptation to changes in preload. It follows from the Laplace law that in a thin-walled sphere, an increase in intraluminal pressure results in an increase in wall stress, unless the thickness of the chamber wall is augmented or the internal radius of the chamber is reduced (Fig. 3.6). An important adaptation of the RV to the high pressure in PAH is to increase wall thickness by accumulating muscle mass (hypertrophy) and to assume a more rounded shape. As indicated in Fig. 3.7, the RV wall stress is low in normally functioning ventricles and increases in severe systolic dysfunction.

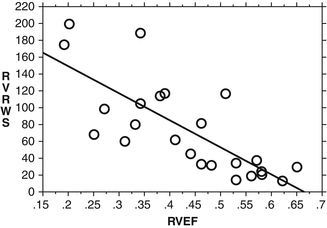
Fig. 3.6
Laplace’s law: σ = (Ρ x r)/h, where σ = wall stress, P = pressure, r = internal radius, and h = wall thickness. RV under normal conditions (a) and in PAH (b). PAH is characterized by increased RV wall stress due to increased pressure and a large radius. As an adaptive mechanism to increased wall stress, wall thickness subsequently increases leading to RV hypertrophy
Fig. 3.7
Inverse relationship between a calculated measure of end-systolic circumferential right ventricular wall stress (RVWS) and right ventricular systolic function as measured by ejection fraction (RVEF). Right ventricular wall stress summarizes the major factors that contribute to wall stress on the right ventricle including pressure, dilation or radius, and wall thickness (Adapted from Voelkel et al. [33])
Adaptive remodeling is characterized by concentric pattern (higher mass-to-volume ratio) and preserved systolic and diastolic function (e.g., ventricular remodeling observed in patients with Eisenmenger syndrome). The increase in ventricular mass induced by an increase in afterload is predominantly the result of protein synthesis and an increase in cell size through the addition of sarcomeres [34]. Proliferation of cardiomyocytes parallels extracellular matrix (ECM) synthesis. Alterations in the ECM, which regulates the milieu for electrical propagation, predispose to conduction abnormalities and arrhythmias [35].
3.2.2 The RV Under Volume Overload
One of the major characteristics of the RV is that it tolerates volume overload better than pressure overload and, therefore, may remain well-adapted to right-sided valvular regurgitant lesions for extended periods of time [36]. This explains why patients with PAH caused by left-to-right shunt remain asymptomatic, until severe pulmonary vasculopathy develops, creating ultimately a reversed shunt (Eisenmenger’s physiology). Even after Eisenmenger’s physiology is well established, the clinical condition of these patients is better than for patients with idiopathic PAH, due to preconditioning by the prior volume load or retention of fetal right heart phenotype characteristics [37, 38].
3.2.3 Maladaptive Hypertrophy: Transition from Adaptive Hypertrophy to Dilatation and Failure
The RV is not capable to sustain long-term pressure overload. Though the RV is highly efficient, it is ill adapted to sudden increases in afterload. When presented with an acute increase in afterload, cardiac output is likely preserved through an increase in RV end-diastolic volume via a “Frank–Starling mechanism” as well as a homeometric mechanism that is characterized by an increase in work and more rapid development of pressure at a given end-diastolic volume [39, 40]. RV dilatation leads to increased wall tension that subsequently increases myocardial oxygen demand. At the same time, RV perfusion decreases and a vicious circle of further compromised contractility and dilatation ensues. This type of maladaptive remodeling is associated with more eccentric hypertrophy, as opposed to concentric hypertrophy in adaptive remodeling, and worse systolic and diastolic function. This pathophysiology is particularly evident in patients with PAH [41, 42] as illustrated in Fig. 3.8.
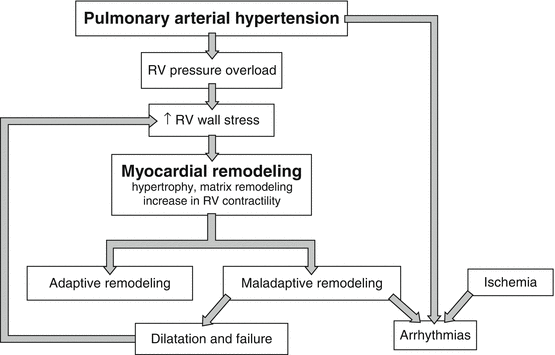
In PAH, the RV free wall is still contracting while the LV is already in its early diastolic phase, leading to late systolic leftward shift of the interventricular septum [44]. Because myocytes under mechanical stress prolong their contraction time and action potential duration, right-to-left ventricular dyssynchrony will increase in the failing RV with increasing wall stress. Common clinical conditions encountered in certain patient populations (tricuspid regurgitation due to annular dilatation, right-to-left shunt due to patent foramen ovale, septal defect, or atrial septostomy) further aggravate maladaptive remodeling, leading to more severe RV dysfunction.
When studying ventricular remodeling due to pressure overload, several differences can be noted between the right and left ventricles. An important feature is that RV enlargement occurs much earlier in the course of PAH compared with the pressure-overloaded LV (e.g., in aortic stenosis). This can be partially explained due to increased RV wall stress for a comparable pressure increase. This is attributed to less thickness of the RV as compared to LV [43]. As indicated in Fig. 3.9, the magnitude of pressure overload is much higher in PAH (340 % vs. 180 %) compared with that of LV pressure overload (180 %) [45, 46]. In contrast, mass index, as a surrogate marker of hypertrophy, is higher in LV pressure overload than in RV pressure overload. This results in normalization of LV wall stress, whereas this is not the case for RV. This implies that the mechanisms leading to LV failure do not extrapolate to RV failure [47].
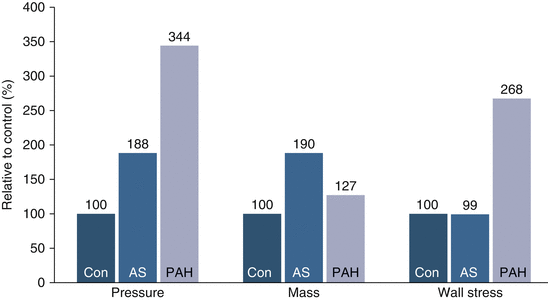
Fig. 3.9
Magnitude of pressure, ventricular mass, and wall stress increase of the affected ventricle in control patients (Con) and patients with aortic stenosis (AS) and pulmonary arterial hypertension (PAH). A marked increase in relative pressure and wall stress is evidenced in patients with PAH that is not anticipated with relative increase in ventricular mass (Adapted from [45, 46])
Regarding fibrosis of the RV in maladaptive physiology, this is much less extensive in patients with RV pressure overload (often <10 % of ventricular volume) and most often limited to the interventricular septum as compared to myocardial fibrosis in LV hypertrophy [48, 49]. In clinical terms this explains recovery of the right heart failure in the majority of patients with severe PAH after lung transplantation [50, 51]. Ongoing research aims to identify which subpopulation of patients with PAH would not recover after lung transplantation alone and would, therefore, benefit from heart-lung transplantation [52]. Table 3.3 summarizes the spectrum of adaptive versus maladaptive remodeling of the RV.
Characteristic | Adaptive remodeling | Maladaptive remodeling |
|---|---|---|
RV size | Normal to mild dilatation | RV enlargement |
Mass/volume ratio | Increased | Reduced |
RVEF | Normal to mildly decreased | Decreased |
Perfusion | Normal or mildly impaired | Decreased |
Metabolism | Normal glucose uptake | Increased glucose uptake |
3.2.4 Pathophysiology of RV Ischemia
The RV is supplied predominantly by the right coronary artery (RCA) in a right-dominant circulation (approx. 75 % of population) [53]. Proximal RCA flow, in normal hemodynamic conditions, is maintained during both systole and diastole [54]. At the distal RV myocardial territories, beyond the RV marginal branches, diastolic coronary blood flow predominates. Acute RCA occlusions proximal to the right ventricular branches compromise RV free wall perfusion, resulting in RV dysfunction in nearly 50 % of patients with transmural inferoposterior myocardial infarctions [55]. It should be noted that the infundibulum receives its blood supply from the conal artery, which has a separate ostial origin in 30 % of cases. The separate ostium explains the preservation of infundibular contraction in the presence of proximal RCA occlusion, which is important for maintaining RV cardiac output [5].
An important characteristic of the RV is that it shows a relative resistance to irreversible ischemic injury, which is mainly attributed to lower oxygen demand/supply balance (Table 3.4) and to the more extensive collateral system of this ventricle [56], especially through the moderator band artery of the RV which originates from the first septal branch of the left anterior descending artery [4]. This resistance of the RV to irreversible ischemic injury leads to significant RVEF increase during the recovery period [57]. Thus, myocardial stunning plays an important role after RV infarction, and adequate support (either pharmaceutical or mechanical) of the RV for a period of time is crucial for a favorable outcome.
Table 3.4
Characteristics of RV oxygen demand/supply balance
Lower oxygen requirements |
Lower resting coronary blood flow |
Higher systolic contribution to total perfusion |
Greater effect of flow and pressure on oxygen demand |
Less coupling between flow and contractile function |
Greater oxygen extraction reserve |
Higher dependency on NO as a regulator of blood flow |
Pronounced α-adrenergic vasoconstriction |
Similar reduction in subepicardial and subendocardial flow during hypoperfusion |
RV ischemia is a prominent feature in patients with RV hypertrophy [58]. Three potential mechanisms for this phenomenon have been suggested not mutually exclusive [59]: (1) increased wall tension increases oxygen consumption and decreases oxygen supply by compression of the coronary circulation, (2) RCA perfusion pressure (Ao-RV pressure difference) is reduced, and (3) the loss of small vessels (arteriolar–capillary rarefaction), rather than reduced coronary perfusion pressure. Van Wolferen et al. found that the mean RCA flow is decreased in severe RV hypertrophy and is directly correlated with RV systolic pressure and RV mass [60]. RV contractility is maintained until perfusion pressures fall below 50 mmHg [61]. It remains controversial whether reduced epicardial coronary perfusion pressure or microvascular ischemia (capillary rarefaction) is the cause of ischemia that elicits the metabolic switch that ultimately leads to RV failure [5]. Whether microvessels in this setting vanish or whether angiogenesis simply lags behind the degree of hypertrophy remains elusive.
3.2.5 Pathophysiology of the RV in Congenital Heart Disease (CHD)
In patients with CHD, the same pathophysiologic principles responsible for development of RV failure apply. However, in this setting, specific additional anatomical conditions [i.e., whether the anatomic RV supports the pulmonary circulation (subpulmonary RV) or the systemic circulation (systemic RV)] may further complicate the pathophysiologic process. In clinical terms RV failure is closely related to patients’ outcome [62]. During the last 50 years, evidence is accumulating that RV dysfunction leads to considerable morbidity and mortality. Therefore, RV function in certain groups of CHD patients needs close surveillance and timely and appropriate intervention to optimize outcomes.
In patients with left-to-right shunting [as in large atrial septal defect (ASD)], there is significant volume overload and remodeling of the RV, which is well tolerated for a long period before developing symptoms. Reverse remodeling occurs after off-loading the ventricle (i.e., surgical closure of the shunt). This phenomenon is more pronounced with decreasing age [63]. Moreover, volume overload is the primary form for RV failure in conditions where there is hypoplastic RV chamber (either structural or functional, such as in Epstein anomaly). In these conditions the RV chamber is incapable of adequately handling the systemic venous return and ultimately fails.
Another mechanism of RV dysfunction in CHD is RV dilatation that occurs secondary to severe pulmonary regurgitation, such as in patients with repaired tetralogy of Fallot (TOF). This condition is associated clinically with decreased exercise tolerance and atrial and ventricular arrhythmias and, hence, carries a worse prognosis [62]. Pulmonary valve replacement in this setting results in ventricular reverse remodeling with a decrease in RV volume. Another pattern of RV failure, which is called “restrictive RV physiology,” is present in conditions as occur in some patients after repair of TOF. It is defined by the presence of forward and laminar late diastolic pulmonary flow throughout respiration [62], and it is associated with a low cardiac output state [64]. In this setting a less compliant RV, late after TOF repair, counteracts the effects of chronic pulmonary regurgitation and results in a smaller RV and a shorter QRS duration.
Another pathophysiologic mechanism leading to RV failure in CHD is RV outflow tract obstruction (such as in pulmonary valve stenosis, double-chambered RV, infundibular hypertrophy, or dynamic obstruction of the RV outflow tract). An important feature of the RV is that it adapts well even to severe outflow obstruction with RV hypertrophy, without producing symptoms. Eventually, disease progression may lead to severe tricuspid regurgitation and clinically evident RV failure [62].
Severe pressure overload of the RV may also result when the anatomic RV supports the systemic circulation [systemic RV, such as in patients with D-transposition of the great arteries (TGA) who underwent atrial switch surgery as well as in patients with congenitally corrected L-TGA] [65]. In this condition impaired myocardial perfusion, uncoordinated myocardial contraction, as well as systemic atrioventricular valve (tricuspid valve) regurgitation result in late RV failure.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


