Fig. 6.1
Dynamics of energy transformation in coupled flows, I, and forces, F, of aerobic metabolism and energy utilization. 2[H] indicates the reduced hydrogen equivalents of carbon substrates with electron transfer to molecular oxygen. FO2 is the Gibbs force or molar Gibbs energy of oxygen consumption (typically −450 kJ/mol O2). H+ in to H+ out is the process of proton translocation across the inner mitochondrial membrane, generating a proton motive force, FH+ = Δp which has a chemical and electrical component (proton gradient and membrane potential). A short circuit (H+ leak) dissipates the energy of the translocated protons, which otherwise is used to phosphorylate ADP to ATP, with an output force of 52–66 kJ/mol ATP under intracellular conditions (Gnaiger 1993). The efficiency of coupling is diminished by potential proton slips of the proton-energy conserving pumps (Complexes CI, CIII, and CIV) and the proton-slip of proton-energy utilizing pump (ATP synthase or CV). Production of reactive oxygen species (ROS) is another component reducing the coupling between oxygen flux and ATP turnover. ATPase activity may short-circuit the ATP cycle and dissipate the energy stored in ATP, which otherwise is used to drive various output flows, from biosynthetic pathways, ion pumping to muscle contraction
As physical activity is diminished despite sufficient energy supply, the electron transport system becomes more reduced (2[H]), the electrical potential across the inner mitochondrial membrane is increased, and electron leak to oxygen increases the generation of reactive oxygen species (ROS; Fig. 6.1). Under these conditions of diminished energy utilization, reduction of mitochondrial density may be considered adaptive (Fig. 6.2). At low mitochondrial density and tissue mass-specific OXPHOS capacity, the work to be done per mitochondrial unit in the tissue is increased, keeping hyperpolarization in check. Simultaneously, the density is decreased of mitochondrial ROS production sites (Complexes I and III). The respiratory LEAK/OXPHOS flux control ratio is diminished in untrained healthy and obese subjects (Pesta et al. 2011; Hey-Mogensen et al. 2010), thus increasing the proton leak and maintaining the proton-motive force lower at rest (Fig. 6.1). Importantly, however, healthy mitochondria act not only as ROS producers but also as ROS scavengers (particularly CoQ and cytochrome c); mitochondria are important redox regulators, and these cell-signaling functions are progressively lost as mitochondrial density declines. Below a threshold level of low mitochondrial density, the adaptive range for downregulation of mitochondrial biogenesis is exceeded. This is the transition to a proinflammatory state (“mitochondrial fever”), putting the cell at risk of further degeneration and apoptosis, with all sequelae of age-related diseases.
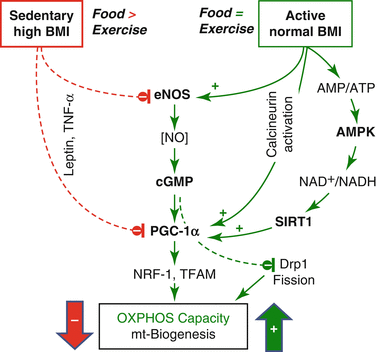
Fig. 6.2
Central role of peroxisome proliferator-activated receptor γ coactivator-1α (PGC–1α) in the control of tissue OXPHOS capacity and mitochondrial (mt) biogenesis. Calcineurin (Lin et al. 2002), eNOS (Nisoli et al. 2007), and AMP-activated protein kinase (AMPK) (Cantó et al. 2009) increase PGC-1α expression, which in turn increases coordinated expression of nuclear DNA and mtDNA through downstream transcription factors, nuclear respiratory factor 1 (NRF-1), and mt transcription factor A (TFAM). PGC-1α expression is inhibited by the proinflammatory cytokine tumor necrosis factor α (TNF-α) and high levels of leptin. At a given mt volume density as a net result of mt biogenesis and mt degradation, NO exerts an additional role on OXPHOS and muscular repair by elongation of tubular mitochondria through a reduction of fission by inhibition of dynamin-related protein (Drp1) (De Palma et al. 2010). In normal-weight animals, moderate caloric restriction enhances OXPHOS capacity by increasing eNOS expression (Nisoli et al. 2005)
In rats artificially selected for low aerobic capacity, impaired regulation of oxidative pathways (PGC-1α; Fig. 6.2) in skeletal muscle precedes the increase in body weight and is linked to a high incidence of cardiovascular disease (Wisløff et al. 2005). In this model of the metabolic syndrome, high-intensity exercise training is superior to moderate-intensity exercise for ameliorating cardiovascular risks (Haram et al. 2009). Selective breeding for high running capacity, in turn, increases OXPHOS capacity and enhances ROS production, whereas oxidative DNA damage is decreased due to a compensatory increase in antioxidant capacity (Tweedie et al. 2011). A high-fat diet increases mitochondrial H2O2 production and decreases redox-buffering capacity (Anderson et al. 2009). While mitochondrial oxidative stress mediates heart failure (Dai et al. 2011), endurance exercise increases antioxidant enzyme activity in cardiac mitochondria, attenuates ROS-induced cytochrome c release, and reduces the threshold for ROS-induced apoptosis (Kavazis et al. 2008). Chronic heart failure (CHF) in humans is associated with a decreased OXPHOS capacity particularly for fatty acid oxidation (Lemieux et al. 2011). In vastus lateralis of CHF patients, OXPHOS capacity (with glutamate+malate as substrates) and the positive response to training are preserved with respect to sedentary controls (Zoll et al. 2003; Garnier et al. 2005). Stimulation of mitochondrial biogenesis entails beneficial effects on various levels, including impaired carcinogenesis (Wang and Moraes 2011).
6.6 Direct Effects of Physical Activity on the Cardiovascular System
Whereas several decades ago physicians recommended prolonged rest for patients with heart diseases, nowadays regular physical activity or even vigorous exercise is an evidence-based part of the prevention and therapy of various heart diseases (Shephard and Balady 1999). “To date, the only practical and sustainable countermeasure capable of promoting cardioprotection is regular bouts of endurance exercise” (Kavazis et al. 2008), reducing the risk of death due to ischemia-reperfusion in humans (Ignarro et al. 2007) and animal models (Powers et al. 2002). The favorable effects of exercise are partly due to the modification of cardiovascular risk factors but also to direct effects on the heart, i.e., cellular and molecular remodeling (Ellison et al. 2012). Exercise-induced cardiac hypertrophy, a balanced increase of left ventricular mass accompanied by neoangiogenesis, represents the most widely known structural adaptation of the heart to regular endurance exercise (Weiner and Baggish 2012). In contrast to the old paradigm that the heart represents a post-mitotic organ without regenerative capacity (Ellison et al. 2012), it has recently been demonstrated that 50 % of cardiomyocytes are exchanged during a normal human life (Bergmann et al. 2009). Waring et al. (2010) reported an elevated proliferation, number, and cardiogenic differentiation of endogenous cardiac stem and progenitor cells following controlled endurance training. Animal studies indicated that exercise-induced cardiomyocyte hypertrophy and renewal depended on a reduced expression of the transcription factor C/EBPb and an increase in the expression of CBP/p300-interacting transactivator (Boström et al. 2010). Also functional benefits like improved contractility have been shown in unloaded cardiomyocytes after endurance training, probably due to a modification of intracellular handling of Ca2+ (Ellison et al. 2012). Additionally, regular endurance exercise increases the number and size of coronary vessels, endothelial function, coronary blood flow, oxygen transport capacity, and oxygen extraction (Duncker and Bache 2008; Ellison et al. 2012). These adaptations are triggered by signals including hemodynamic forces like flow and stretch, tissue ischemia, and growth factors like vascular endothelial growth factor (VEGF) and fibroblast growth factor-2 (FGF-2) (Ellison et al. 2012). Exercise is also an important regulator of eNOS-mediated vascular release of NO (Balligand et al. 2009) which is closely associated with cardiovascular remodeling, improved Ca2+ handling and endothelial function, and mitochondrial biogenesis (Fig. 6.2).
6.6.1 Endothelial Dysfunction
Blunted endothelium-dependent vasodilatation is an early sign in patients with risk factors for CVD (McVeigh et al. 1992). This dysfunction of the endothelium is typically caused by a reduction of NO bioactivity. NO inactivation is caused by excessive ROS production and has been demonstrated in patients with risk factors for CVD (Cai and Harrison 2000). There are several lines of evidence supporting the potential of regular physical activity to prevent or reverse endothelial dysfunction, e.g., in patients with coronary artery disease or T2DM (Szostak and Laurant 2011). This becomes evident since exercise training represents one of the most important regulators of eNOS-mediated release of NO from the endothelium (Indolfi et al. 2002). NO release results at least partly from the increased vascular shear stress during exercise (Tuteja et al. 2004). Elevated NO production not only favors vasorelaxation but also reduces platelet adhesion and aggregation thereby eliciting its beneficial effects in patients with coronary artery disease (Tuteja et al. 2004). Exercise training represents an antioxidant therapy thereby increasing NO bioavailability and improving endothelial function (Di Francescomarino et al. 2009). Both reduced NO bioactivity and oxidative stress seem to contribute to chronic systemic inflammation and endothelial dysfunction (Szostak and Laurant 2011; Rizvi 2009) which seems to be improved by regular exercise training (Petersen and Pedersen 2005).
6.7 Primary Preventive and Therapeutic Targets of Physical Activity
Oxidative stress, low NO bioavailability, and inflammation are considered as primary targets for modification of risk factors by regular exercise training. All these factors are closely interrelated and may play important roles in the development of traditional risk factors and atherosclerosis.
ROS inactivate NO resulting in endothelial dysfunction, promote LDL oxidation, and the progression of atherosclerosis (Davignon and Ganz 2004). During acute exercise the mitochondrial electron transfer system contributes to an elevation of ROS production (Bloomer 2008). Concomitantly, there is an increase of the pro-oxidant enzymes which is associated with ROS markers like F2-isoprostanes (Brennan et al. 2003). NADPH oxidase activity is closely related to ROS generation and has been shown to be associated to physical inactivity (Szostak and Laurant 2011). During regular and chronic exercise training, antioxidant enzymes increase, e.g., superoxide dismutase (SOD) and glutathione peroxidase, resulting in a more powerful oxidant handling (Stapleton et al. 2010). Consequently, oxidative stress markers like F2-isoprostane decrease (Seals et al. 2008).
Whereas NO production by vasoactive agonists is related to the increase of calcium concentration in endothelial cells, fluid shear stress during exercise activates eNOS by phosphorylation of the serine/threonine protein kinase Akt/protein kinase B in a phosphoinositide 3-kinase-dependent manner (Wyatt et al. 2004). Oxidative stress, caused by an imbalance between oxidant and anti-oxidant enzymes, inactivates NO and favors low NO bioavailability (Szostak and Laurant 2011). The consequences of decreased NO bioavailability include upregulation of endothelin-1 (ET-1) and activation of platelets and coagulation pathways and thus endothelial dysfunction. On the other hand, regular exercise increases NO bioavailability directly by eNOS activation and indirectly by diminishing oxidative stress thereby improving endothelial function (Goto et al. 2003; Szostak and Laurant 2011).
Thus, exercise increases antiatherogenic markers like eNOS and SODs and decreases the expression of ET-1, adhesion molecules like VCAM-1, and ROS as markers of a proatherogenic endothelial cell phenotype (Laughlin et al. 2008) (Fig. 6.3).
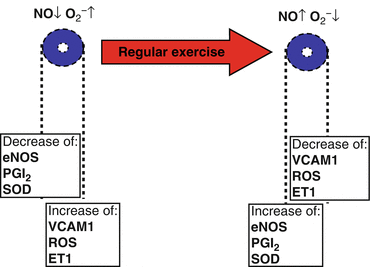
Fig. 6.3
Regular exercise increases antiatherogenic markers like endothelial nitric oxide synthase (eNOS); prostacyclin (PGI2), and superoxide dismutase (SOD) and decreases the expression of endothelin-1 (ET-1), adhesion molecules like VCAM-1, and reactive oxygen species (ROS) as markers of a proatherogenic endothelial cell phenotype (Laughlin et al. 2008)
Both elevated ROS production and decreased NO bioactivity seem to be causally linked to chronic inflammation representing a pathogenic feature of atherosclerosis (Ross 1999). For example, ROS and ROS products activate the nuclear factor κB (NFκB) which contributes to the regulation of proinflammatory genes. NFκB seems to be activated during healthy and diseased conditions. It seems reasonable that the severity and the frequency of stress determine whether responses are favorable or, with inadequate recovery, rather detrimental. Thus, regular exercise with sufficient recovery will promote positive adaptations making the organism more resistant to future perturbations (Kramer and Goodyear 2007) (Fig. 6.4).
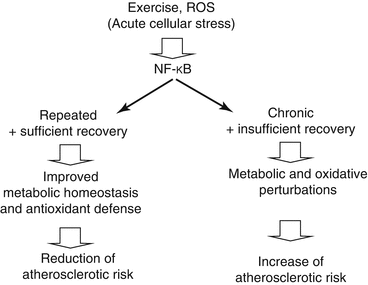
Fig. 6.4
Nuclear factor κB (NFκB) is activated by acute cellular stress. When NFκB is chronically activated in diseases like diabetes, the related metabolic and oxidative perturbations increase the atherosclerotic risk. However, repeated NFκB activation with exercise (and sufficient recovery) improves metabolic homeostasis and antioxidant defense resulting in reduction of the atherosclerotic risk (Kramer and Goodyear 2007)
NFκB has also been suggested to be involved in alterations of fuel metabolism during and following exercise by increasing transcription of the IL-6 gene (Kramer and Goodyear 2007). IL-6 is produced by the contracting skeletal muscle and favors glucose transport and lipid oxidation (Pedersen and Steensberg 2002) and stimulates satellite cell proliferation (Toth et al. 2011). On the other hand IL-6 diminishes tumor necrosis factor-alpha (TNF-α) release. TNF-α is known to promote vascular inflammation, oxidative stress, and reduced NO bioactivity resulting in endothelial dysfunction (Szostak and Laurant 2011) and arrest of mitochondrial biosynthesis (Fig. 6.2). TNF-α also contributes to insulin resistance by increasing lipolysis and the related release of nonesterified fatty acid (NEFA, Plomgaard et al. 2005). Regular physical activity, however, is associated with decreasing concentrations of proinflammatory markers like TNF-α and improved insulin sensitivity thereby inhibiting atherogenesis (Pedersen et al. 2007) (Fig. 6.5).
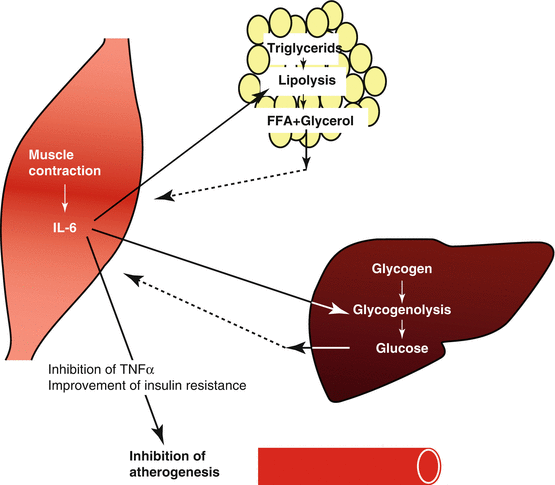
Fig. 6.5
Interleukin-6 (IL-6) is produced by the contracting skeletal muscle and favors glucose transport and lipid oxidation. Regular physical activity is also associated with decreasing concentrations of proinflammatory markers like tumor necrosis factor-alpha (TNF-α) and improved insulin sensitivity thereby inhibiting atherogenesis (Pedersen et al. 2007)
6.8 Summary and Perspectives
In recent years, many studies have focused on the pathophysiological pathways associated with the involvement of oxidative stress and inflammation in the development of metabolic disorders and cardiovascular diseases and the preventive impact of regular exercise training. Ample evidence has been provided that regular exercise increases anti-inflammatory environment, improves anti-oxidant defense, and reduces oxidative stress thereby counteracting the development of atherosclerosis. Nevertheless, much more research is required to provide a deeper insight in cellular and molecular mechanisms explaining the effects of exercise on oxidative stress effects and inflammatory pathways. Findings from this research will help to establish optimal training recommendations in order to maximize the benefits of exercise training for healthy as well as diseased subjects on an individual basis.
Acknowledgments
This chapter is a contribution to K-Regio project MitoCom Tyrol, funded by the Tyrolian Government and the European Regional Development Fund.
References
Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119:573–581PubMedCrossRef
Blair SN, Kohl HW, Barlow CE, Paffenbarger RS, Gibbons LW, Macera CA (1995) Changes in physical fitness and all-cause mortality: a prospective study in healthy and unhealthy men. JAMA 273:1093–1098PubMedCrossRef
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


