Fig. 7.1
Etiology of ARVF categorized according to underlying pathophysiology (ARDS acute respiratory distress syndrome, HTx heart transplantation, PAH pulmonary arterial hypertension, RV right ventricle)
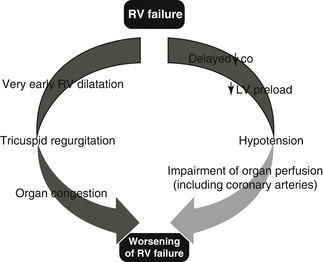
Fig. 7.2
Vicious cycle of autoaggravation that characterizes pathophysiology of RV failure [2] (CO cardiac output, LV left ventricle)
The general principles that apply in the treatment of patients with advanced RV failure are described schematically in Fig. 7.3. A stepwise pharmacologic approach is based on the following principles: optimize preload, reduce afterload, and improve contractility. The main objective is to restore RV function to the point where the patient can either be stabilized for definitive treatment with traditional oral or parenteral pulmonary vasodilators or undergo heart, lung, or heart-lung transplantation. To achieve this end, focus of treatment should be to search for and address reversible causes, reduce RV wall tension, restore RV output, and reduce the adverse influence of a dilated, pressurized RV on LV filling (Table 7.1) [3]. For example, patients with chronic lung disease, such as severe emphysema, causing cor pulmonale, can develop ARVF after infection and, hence, become septic. In this situation, the underlying condition responsible for the chronic RVF cannot be reversed, and management should be directed toward optimizing RV function. In another setting, ARVF is the result of a sudden increase in RV afterload, such as after acute pulmonary arterial hypertension (PAH) due to massive pulmonary embolism. In this situation, the first priority should be to relieve the increase in afterload with thrombolysis or pulmonary thromboendarterectomy.
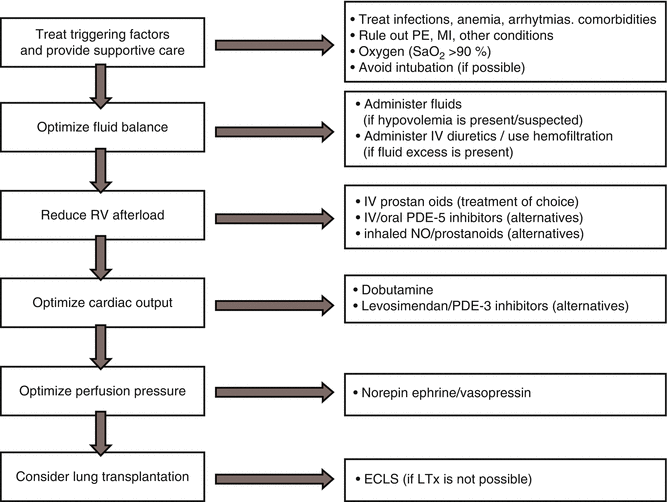
Fig. 7.3
Algorithm for the management of RV failure in the critically ill patient (ECLS extracorporeal life support, LTx lung transplantation, MI myocardial infarction, NO nitric oxide, PDE phosphodiesterase, PE pulmonary embolism)
Table 7.1
Etiologic therapies in acute right ventricular failure
Postcardiotomy compression of PA or graft failure |
Surgical correction |
Pulmonary embolism |
Thrombolysis |
Anticoagulation |
Pulmonary thromboendarterectomy |
RV myocardial infarction |
Thrombolysis |
PCI |
Coronary artery surgery |
Sepsis |
Antibiotics |
Pulmonary arterial hypertension |
Inhaled NO |
Prostaglandins |
Sildenafil |
Endocarditis |
Antibiotics |
Surgery when indicated |
LV dysfunction |
Optimization of LV function |
Congenital heart disease |
Conservative management |
Surgical correction |
7.2 Oxygenation and Ventilation Management
Careful management of oxygenation is essential to the treatment of RVF. Hypoxemia is present in severely compromised patients with chronic lung disease due to pulmonary-to-systemic shunt associated with ventilation-perfusion mismatch [4, 5]. Anemia further impairs tissue oxygenation and should be treated in order to ensure adequate oxygen delivery to the systemic circulation. Both hypoxia and hypercapnia promote pulmonary vasoconstriction and increase RV afterload [6, 7]. For patients with refractory hypoxemia, continuous positive airway pressure or noninvasive ventilation should be considered before intubation. In case that mechanical ventilation is deemed mandatory, hypotension and loss of RV contractility must be prevented, and the infusion of catecholamines before anesthesia should be considered. Etomidate and ketamine are the preferred drugs for induction of general anesthesia given their relatively beneficial hemodynamic profile with a low negative inotropic effect [8, 9].
In patients on mechanical ventilation, large tidal volumes, high peak inspiratory pressure, and high positive end-expiratory pressure further increase pulmonary vascular resistance (PVR) and, thus, RV afterload. To prevent excessive increase in RV afterload, the ventilator strategy should aim in achieving normal PaO2 and mild hypocapnia with moderate tidal volumes (~8 ml/kg), low levels of positive end-expiratory pressure (<12 cmH2O), and maintenance of peak inspiratory pressure of <30 mmHg [10]. In order to control peak inspiratory pressure at the desired level without increasing tidal volume, the ventilator mode is set to pressure-controlled inverse ratio ventilation (PCIRV). Using this type of ventilation, a relatively higher respiratory rate and concentration of inspired oxygen may be required. Less common ventilator techniques used primarily in patients with acute respiratory distress syndrome (ARDS), such as high-frequency oscillatory mode and prone positioning, have shown mild improvement in RV function in small clinical studies [11, 12].
7.3 Optimization of RV Preload
Optimal fluid management is critical for successful treatment of the failing RV. Evaluation of optimal intravascular volume status in patients with compromised RV is problematic [13]. In the early stages of critical illnesses, intravascular volume can fall rapidly in response to increased vascular permeability and insensible losses. Medication administered, mainly sedatives and analgesics, blunts sympathetic vasoconstriction of the systemic venous circulation, leading to decreased venous tone and reduced right-sided return. Mechanical positive-pressure ventilation leads to an increase in intrathoracic pressure which impedes RV preload by reducing RV transmural filling pressure.
In order to optimize cardiac output in patients with ARVF, maintenance of adequate right-sided filling pressure is essential [14]. It is important to understand that RV preload requirements differ substantially between individuals depending on whether afterload is normal or increased. As shown in Fig. 7.4, the RV has a flatter Starling curve compared to the LV, meaning that RV contractility increases minimally over a wide range of filling pressures. Hence, a considerable amount of volume unloading may be necessary before any improvement in RV function is seen.
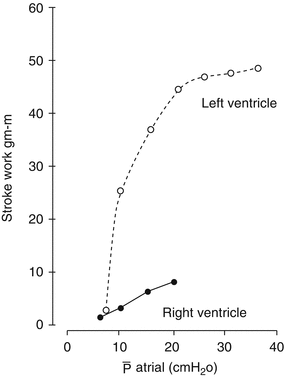
Fig. 7.4
Difference in response of LV and RV after an increase in preload. The RV curve is flattened indicating that higher amount of fluid is required in order to increase preload (Adapted from Braunwald [15])
When RVF occurs in the setting of normal PVR, such as after right-sided myocardial infarction or in the postcardiotomy setting, RV end-diastolic pressure often needs to be increased above normal levels to maintain adequate cardiac output. However, when RVF occurs in the setting of increased RV afterload, excessive volume administration can be detrimental. Excessive RV volume loading leads to displacement of the interventricular septum toward the LV, which subsequently impairs LV diastolic filling (ventricular interdependence; see Chap. 3) [13]. Moreover, tricuspid regurgitation due to annular dilation may further increase right-sided filling pressures, while at the same time increased RV wall tension leads to an increased myocardial oxygen demand and decreased RV perfusion. In this setting, intravascular volume needs to be decreased considerably with administration of diuretics.
Optimal right-sided filling pressure may vary considerably between individual patients based on RV contractility and afterload. In general, RV transmural filling pressure (central venous pressure, CVP) should be maintained in a moderately elevated range, typically 8–12 mmHg, and then adjusted to optimize RV function and cardiac output. Once CVP has risen above 10–14 mmHg, further volume loading is usually unfavorable [16]. Erroneous management of RV failure in the ICU involves volume loading for the treatment of elevated creatinine. In fact, recent evidence suggests that venous congestion, rather than reduced cardiac output, is the major contributor to renal dysfunction in this setting (cardiorenal syndrome) [17]. Thus, reduction rather than elevation of CVP may improve renal perfusion [1]. This is the rationale for ultrafiltration use as therapeutic option in refractory to diuretic therapy cases [18].
7.4 Pharmacologic Aspects of Loop Diuretics
Loop diuretics inhibit Na+, K+, Cl−, Mg2+, and Ca2+ reabsorption in the ascending limb of the loop of Henle [19]. They reach the luminal transport sites through the tubular fluid and are actively secreted into the urine by the cells of the proximal tubule. Approximately 50 % of furosemide dose is excreted unchanged into the urine, while the remaining 50 % is conjugated to glucuronic acid in the kidneys. This explains prolonged half-life of furosemide in patients with renal insufficiency [20]. In contrast to furosemide, bumetanide and torsemide are metabolized by the liver and do not show prolonged half-lives in renal insufficiency. The plasma half-life is approximately 1.5–2 h while for bumetanide is lower (1 h) [21]. This determines the frequency of intravenous diuretic administration. This knowledge is of particular importance for the management of ARVF, where renal insufficiency is common.
It is important to consider that with decreasing renal function, the amount of loop diuretic secreted into the tubular fluid is more than five times smaller compared to healthy individuals [20]. Thus, larger dose is required to attain an effective diuretic response. In general, the maximal natriuretic response in heart failure occurs with intravenous bolus doses of 160–200 mg of furosemide and the equivalent doses of bumetanide (8–10 mg). In case of poor response to intermittent doses of loop diuretics, continuous intravenous infusion can be initiated. This rate of continuous infusion is determined by the patient’s renal function (Table 7.2).
Table 7.2
Doses for continuous intravenous infusion of loop diuretics
Diuretic | Loading (mg) | Infusion rate (mg/h) | ||
|---|---|---|---|---|
CrCl <25 ml/min | CrCl 25–75 ml/min | CrCl >75 ml/min | ||
Furosemide | 40 | 20–40 | 10–20 | 10 |
Bumetanide | 1 | 1–2 | 0.5–1 | 0.5 |
Torsemide | 20 | 10–20 | 5–10 | 5 |
Intravenous-administered loop diuretics reduce LV end-diastolic volume and increase venous capacitance within 5 min, before any recognizable effect in urine output. The acute vasodilatory effect is mediated by prostaglandin E2 (PGE2) [22]. With progressive diuresis, pulmonary arterial pressure is reduced. Cardiac index and other load-dependent indices of cardiac performance are improved after diuresis due to a decreased afterload of the pulmonary and systemic circulation, before a decrease in preload [23]. By reducing intravascular volume, LV and RV filling pressures decrease resulting in wall stress decrease, which is beneficial in enhancing perfusion of both ventricles. However, excessive reduction of LV filling pressures caused by diuretics should be avoided, as it can lead to a reduction in cardiac output in patients with advanced heart failure. Moreover, diuretic-induced volume contraction is responsible for neurohormonal activation that precipitates renal dysfunction. In patients with heart failure, increased angiotensin II concentration and plasma renin activity is observed that reflect the intensity of diuretic therapy [21]. Renin release is triggered by volume contraction and stimulation of baroreceptors at the macula densa. It has been postulated that the lowest level of neurohormonal activation, as reflected with catecholamines and aldosterone concentration, is the best indicator of optimal volume status. Total body fluid as manifested by peripheral edema, ascites, and sacral edema indicates the need for additional diuresis. Optimization of total body fluid overload contributes to a reduction in rehospitalization rates of patients presenting with acutely decompensated heart failure [24]. Even though use of diuretics plays a key role in the management of ARVF, limited data regarding their targeted use for primary RVF is dispersed in the literature.
7.5 Optimization of RV Contractility
Impaired RV contractility in patients with ARVF is induced primarily by three interrelated factors: (1) overdilation of the RV free wall, leading to overstretching of cardiomyocytes to a level beyond the positive effect of the Frank-Starling mechanism; (2) derangements in cellular metabolism, leading to decreased myocardial contractile forces; and (3) insufficient oxygen delivery due to decreased coronary artery perfusion [13]. In the critically ill patient, a sudden increase in RV preload and/or afterload increases significantly RV free wall tension and oxygen demand. At the same time, interventricular septum shifts to the left impeding LV filling, thus reducing LV output and decreasing coronary artery pressure. Perfusion of the RV free wall is determined by the difference in RV free wall tension and coronary artery pressure [25]. Systemic hypotension further impairs perfusion of the RV. When fluid resuscitation results in RV distension without increase in systemic arterial pressure, this may aggravate RV ischemia. In this clinical situation, cautious use of vasopressor therapy is required to restore systemic blood pressure to levels above RV systolic pressure [26, 27]. Hemodynamic instability is associated with metabolic derangement, including acid/base disturbances, and generation of reactive oxygen species as well as inflammatory cytokines that impair oxygen utilization at a cellular level [28, 29].
Inotropes increase myocardial contractility. Their use is largely confined to critically ill patients with profound hemodynamic impairment where tissue perfusion is not sufficient to meet metabolic requirements. Even though inotropic support is mainly targeted to the LV, which comprises the greater proportion of myocardial mass, it may improve RV cardiac output. Selection of the optimal inotrope depends on the severity of RV and/or LV dysfunction as well as on the desired effects on systemic and pulmonary vascular tone (Table 7.3), titrated accordingly.
7.6 Dobutamine
Dobutamine acts principally through stimulation of β1-adrenergic receptors, with lesser stimulation of β2– (3:1 ratio) and α-adrenergic receptors [30, 31]. Thus, dobutamine enhances ventricular contraction with little net effect on PVR. Low-dose dobutamine (2–5 μg/kg/min) increases cardiac index and stroke volume and decreases PVR and systemic vascular resistance (SVR) [32, 33]. At higher doses (>10 μg/kg/min), dobutamine causes tachycardia, increased oxygen consumption, increased PVR, and systemic hypotension that exert a detrimental effect on RV function. This is due to the finding that in advanced heart failure, cardiac β1-adrenergic receptors are downregulated due to chronic stimulation from the elevated circulating catecholamines or blocked by β1-blockers. In this setting, most of dobutamine’s hemodynamic effects are mediated through β2-adrenergic stimulation [34].
In case of ARVF, such as after RV myocardial infarction, dobutamine infusion begins at 3–5 μg/kg/min titrated according to its response (1–2 μg/kg/min every 30–120 min). Dose is escalated until the desired clinical or hemodynamic effects are attained; infusion rate of 15 μg/kg/min should be avoided as it is related to a poor outcome in patients with acute heart failure [34]. This results in increased contractility, stroke volume, and cardiac output as well as reduced ventricular filling pressures, SVR, and PVR, hence, improving organ perfusion [35, 36]. Dose titration (starting low and maintaining as low as possible) should aim in obtaining these favorable effects, without significantly increasing heart rate or producing dysrhythmias. It should be emphasized that dobutamine enhances atrioventricular node conduction, and thus, it can accelerate ventricular response in patients with atrial fibrillation or flutter. Moreover, it results in myocardial hypoperfusion and ischemia, when coronary artery disease is present due to increased myocardial oxygen consumption. However, this is not the case in patients with normal coronary arteries, where this effect is balanced by a proportional rise in coronary blood flow and myocardial oxygen delivery [37, 38].
Dobutamine has a short half-life of 2.37 ± 0.7 min in patients with heart failure, due to its rapid metabolism. This mandated continuous intravenous infusion for obtaining adequate bioavailability [39]. Its desired hemodynamic effect is attainable within 10–15 min of dose initiation, and adverse effects vanish in equal time frame from stopping the infusion. Dobutamine is an ideal inotropic agent for patients with RV systolic dysfunction, elevated ventricular filling pressures, reduced stroke volume, and systemic systolic pressures of 70–100 mmHg. Dobutamine due to its predominant effect on contractility represents a first-line inotrope in primary RV dysfunction. It creates a “pharmacologic bridge” to advanced management (e.g., mechanical circulatory support or heart transplantation). Comparative studies proved that it may better enhance hemodynamics when compared to volume loading alone [40], vasodilators, such as with nitroprusside [41], or inopressors, such as norepinephrine [42]. Furthermore, dobutamine is considered a first-line inotrope (usually combined with a vasoconstrictor) due to the marked increase in cardiac output it may produce [43, 44].
7.7 Phosphodiesterace-3 Inhibitors
Phosphodiesterase (PDE) inhibitors decrease cellular cyclic AMP degradation, resulting in elevated levels of cyclic AMP in cardiac and smooth muscle myocytes. The physiologic effect of this mechanism is increased contractility combined with dilation of systemic and pulmonary vessels. Collectively, therefore, PDE inhibitors improve cardiac output by exerting a positive inotropic effect together with decreasing preload and afterload, thus giving rise to the term “inodilator” [32].
The selective PDE3 inhibitors include milrinone, enoximone, and amrinone. Their mechanism of action is mainly by stimulating myocardial contractility and accelerating myocardial relaxation. Moreover, they produce balanced arterial and venous dilation with consequent fall in systemic and pulmonary vascular resistance as well as LV and RV filling pressure. Their arterial and venodilatory effect is greater than dobutamine, while their chronotropic effect is less pronounced. The most frequently used PDE3 inhibitor, milrinone, should be initiated with a loading dose of 50 μg/kg over 15 min followed by continuous infusion of 0.375–0.75 μg/kg/min, titrated accordingly. Half-life of milrinone is 0.5–1 h in healthy individuals, while this is doubled in patients with severe congestive heart failure. It represents the agent of choice among currently available PDE inhibitors for short-term, intravenous inotropic support in patients with preserved mean arterial pressure and RVF secondary to high pulmonary afterload (e.g., pulmonary embolism, PAH due to biventricular failure or following cardiac transplantation) [45]. However, vasodilation-mediated reduction in mean arterial pressure is one practical barrier to milrinone administration in patients with marginal systemic arterial blood pressure, and thus, coadministration with vasopressors is often necessary. Agents such as norepinephrine, phenylephrine, or vasopressin have been used for this purpose [46]. Recently, administration of nebulized milrinone has been used successfully in patients with acute postcardiotomy PAH resulting in reduction in PVR with less systemic hypotension and reduced V/Q mismatch compared with intravenous use [47–49].
7.8 Adrenergic Agonists
Adrenergic agonists (adrenaline, noradrenaline, and dopamine) are not considered first-line inotropes in ARVF due to increased PVR along with increased cardiac index and SVR they generate. Moreover, when administered at higher doses, they may cause tachycardia and precipitate myocardial ischemia. Adrenaline (also known as epinephrine) is the most potent agonist of all adrenoceptors resulting in profound increases in cardiac output and heart rate, mean arterial pressure, and coronary blood flow. At low doses, passive stretch of pulmonary vessels accommodates increases in cardiac output, but as the plasma concentration of adrenaline increases, it eventually increases PVR and hence RV afterload [50]. Moreover, myocardial oxygen demand is significantly increased because of increased heart rate and stroke work, which may be detrimental in cases of RV ischemia. Thus, use of adrenaline should be selected judiciously in patients with severely diminished contractility of the LV and RV, such as following acute myocardial infarction or cardiopulmonary bypass because of its greater stimulation of α-adrenergic receptors [51].
Noradrenaline (also known as norepinephrine) exerts both inotropic and vasopressor effect. Noradrenaline is a potent α-adrenoceptor agonist (vasopressor) and a weaker β2-adrenoceptor agonist (inotrope) [52]. In case of RVF and systemic hypotension due to acute or chronic PAH, it may be useful in maintaining systemic pressure and increasing cardiac index [53, 54]. This is due to increase in SVR more than PVR as found in experimental and clinical studies [55]. Moreover, addition of noradrenaline (0.05–0.5 μg/kg/min) is an established treatment in patients who do not tolerate inodilators due to systemic hypotension.
Dopamine has been found to increase cardiac output in patients with PAH and RV dysfunction, although it may cause a mild tachycardia and increase the PVR/SVR ratio [56, 57]. However, it has less favorable hemodynamic effect in heart failure patients compared to dobutamine [58]. Experimental evidence showed that when dopamine was administered at doses up to 10 μg/kg/min, it did not increase PVR [59]. The same finding was also evident in patients with septic shock who had PAH and RV dysfunction [60].
7.9 Vasopressors
Systemic hypotension in ARVF plays a pernicious role by reducing right coronary artery perfusion precipitating RV myocardial ischemia and, hence, requires treatment with vasopressors. The ideal vasopressor should increase systemic pressure more strongly than pulmonary arterial pressure. Phenylephrine is a pure α1-receptor agonist which may augment coronary artery perfusion; however, it increases PVR and has no impact on RV contractility [27]. Moreover, it can cause reflex bradycardia, which can be troublesome in cases of reduced RV stroke volume. Arginine vasopressin causes systemic vasoconstriction via the vasopressinergic (V1) receptor. It exerts vasodilating properties (including pulmonary vasodilation) at low doses via the release of nitric oxide (NO) as has been demonstrated in experimental studies [63, 64]. This property manifests clinically as a reduction in PVR and PVR/SVR ratio [46, 65]. Thus, it has been extensively used at low doses (0.03–0.067 U/min), in sepsis [66, 67], as well as in patients with RVF and acute PAH with profound hypotension refractory to catecholamines in the postcardiotomy setting [68, 69]. Arginine vasopressin increases diuresis and reduces heart rate, while it is related to fewer tachyarrhythmias in comparison to norepinephrine [66, 70]. However, when it is administered at infusion rates exceeding 0.4 U/min, it becomes detrimental to RV function due to coronary vasoconstriction [71, 72].
Table 7.3
Hemodynamic effects of commonly used inotropes and vasopressors
Agent | CI | PVR | SVR | PVR/SVR |
|---|---|---|---|---|
Dobutamine | ↑↑ | ↓ | ↓ | ↑↓ |
Milrinone | ↑↑ | ↓↓ | ↓↓ | ↓ |
Epinephrine | ↑↑↑ | ↑ | ↑↑ | ↑↓ |
Dopamine | ↑ | ↑ | ↑ | ↑↓ |
Norepinephrine | ↑ | ↑ | ↑↑ | ↑↓ |
Phenylephrine | ↓ | ↑↑ | ↑ | ↑ |
Vasopressin | ↑↓ | ↓ | ↑↑ | ↓ |
7.10 Levosimendan
Levosimendan belongs to the pyridazinone-dinitrile derivative molecule. This drug is defined as a new calcium enhancer with calcium-sensitizing activity. The main mechanism of action is an increase in the troponin C affinity for Ca2+ and a stabilization of the troponin C conformation. It selectively inhibits PDE3, thereby improving diastolic function and myocardial contractility (inotropic effect) without increasing oxygen consumption [73]. It also acts as an arterial and venous vasodilator through calcium desensitization and potassium-ATP channel opening in vascular smooth muscle. This combination of actions endows levosimendan with the properties of both an energy-efficient inotrope and a balanced vasodilator. In addition, levosimendan exerts anti-ischemic and cardioprotective effects via potassium-ATP channels in cardiomyocytes. The most important pharmacokinetic characteristic of levosimendan is that about 5 % of an administered dose is converted by acetylation to an active metabolite (OR-1896), which has a half-life of 77 h, thus allowing cardiovascular effects to persist up to 7–9 days after a 24-h infusion [74].
Intravenous levosimendan is the most comprehensively studied inotropic compound for severe heart failure. The standard dosing regimen of levosimendan consists of a loading dose of 6–24 μg/kg followed by a 24 h infusion at a rate of 0.1–0.2 μg/kg/min. As analyzed in Chap. 12, recent studies which investigated preoperative infusion of levosimendan in patients with borderline RV function planned for LVAD implantation administered the abovementioned dose for a maximum of 48 h without an initial bolus [75]. Repetitive administration of levosimendan has been implemented for optimizing RV function preoperatively before LVAD implantation.
The vast majority of clinical trials have evaluated the effect of levosimendan on clinical outcome of patients with congestive heart failure related to LV dysfunction. There is limited evidence in the literature regarding effectiveness of levosimendan in patients with predominant right heart failure [73]. Levosimendan improves echocardiographic markers of RV systolic function [76]. It also leads to a rapid improvement in hemodynamics, including PVR reduction, as well as significant benefit on RV systolic and diastolic function in patients with decompensated heart failure [77–79]. The same effect applies to patients with RV failure due to ischemic etiology [80, 81] as well as after mitral valve surgery [82]. In the ICU setting, Morelli et al. showed that it improves systemic hemodynamics and regional perfusion compared to dobutamine in septic patients with ARDS [83]. Furthermore, repetitive administration of levosimendan (four times at 2 weeks intervals) proved efficacious in the treatment of patients with chronic PAH of various etiologies without development of tolerance, as shown by Kleber et al. [84]. In general, larger studies are required in order to establish the optimal use of levosimendan in patients with acute or chronic primary right heart failure as well as in patients with increased PVR that are candidates for mechanical circulatory support or heart transplantation.
7.11 Pulmonary Vasodilators
The RV is extremely sensitive to afterload, as discussed in Chap. 3, and its performance could be impaired with even minimal increases in afterload (mean pulmonary artery pressure >25 mmHg) [45]. We may appreciate the importance of RV afterload reduction in right heart function by the rapidity with which RV function is restored after pulmonary endarterectomy and lung transplantation [85, 86]. Thus, selective pulmonary arterial vasodilation is an attractive strategy aiming to improve RV function by reducing PVR without causing systemic hypotension. For this purpose, there are two classes of drugs: (1) intravenous medications with selective effects on the pulmonary vasculature or (2) inhaled agents that are delivered directly to the lungs. Delivery of vasodilators by inhalation may improve ventilation-perfusion (V/Q) mismatch by directing blood flow to ventilated areas of the lung.
The most extensively used inhaled vasodilator is nitric oxide (iNO), which dilates pulmonary vasculature by increasing the production of cyclic guanosine monophosphate (cGMP). Rapid inactivation by hemoglobin in the pulmonary capillaries prevents systemic vasodilation. Effects are limited to ventilated areas of the lung. It, therefore, decreases pulmonary artery pressure and PVR as well as improves oxygenation without increasing intrapulmonary shunt fraction, unlike systemically administered pulmonary vasodilators [87, 88]. Nitric oxide may also improve outcomes in RVF by reducing inflammatory cytokine production in the lung [89]. A synergistic effect can be achieved when combined with systemic inodilators, such as dobutamine, milrinone, or levosimendan [90]. It also provides synergistic pulmonary vasodilation with intravenous or inhaled prostacyclin as well as oral sildenafil [91, 92]. Clinical studies have demonstrated hemodynamic improvements with iNO in patients following RV myocardial infarction, LVAD implantation, or cardiac transplantation [93, 94]. Limitations include accumulation of toxic metabolites (methemoglobinemia, reactive nitrogen species), although without clinical significance. Rebound increase in pulmonary artery pressure that impairs RV function may occur after weaning from iNO, which may be reduced with oral pulmonary vasodilators, such as sildenafil [95, 96]. Even though studies use a mean dose of 20 ppm (range: 10–40 ppm), recent studies advocate a lower dose of iNO (10 ppm), since its response is not dose dependent and the maximum effects on pulmonary vasculature can be achieved at doses as low as 10 ppm [97]. Hence, dose escalation is not beneficial in nonresponders.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


