Fig. 13.1
Changes in brachial and aortic blood pressures, corrected augmentation index, and pulse wave velocity at 5, 10, and 15 min after smoking one cigarette in healthy smokers and nonsmokers. All parameters increased in both groups. Data are presented as mean ± SEM (Reproduced with permission from Mahmud and Feely [10])
The chronic effect of smoking seems to be analogous to that of the acute effect [15, 17–20]. The impact on arterial stiffness appears to be unrelated to the intensity and duration of smoking since small quantities of smoking are able to produce its deleterious effects. The effect of smoking on aortic stiffness may be both BP dependent and BP independent [8]. Interestingly, studies have shown that augmentation index (AIx) and central pressures are higher and pulse pressure is reduced in current and former smokers as compared to never smokers for the same brachial BP levels [10, 19, 21]. The long-term effect of cigarette smoking on the incidence of hypertension has not been elucidated. In two large population studies, current cigarette smoking was independently, yet modestly, associated with an elevated risk of developing hypertension [22, 23]. However, other studies have reported lower BP levels among smokers [24] and increases in BP after smoking cessation [25]. The mildly lower BP in smokers can be attributed to lower body weight [26]. Interestingly, a recent study showed that aerobic exercise may mitigate the harmful effects of smoking since brachial-ankle pulse wave velocity (PWV) values were not different between physically active nonsmokers and physically active smokers [27].
Passive smoking is a well-established risk factor for coronary heart disease [28] with similar detrimental effects on the cardiovascular system to those of active smoking. Passive smoking has immediate and substantial effects on aortic stiffness in smokers [29–31]. These effects are mediated through a nicotine-dependent pathway, impairment of microvascular function, and increase of asymmetrical dimethylarginine [31]. Importantly, data have shown that parental smoking is associated with the development of several cardiovascular risk factors in their children, whereas the exposure to smoking in childhood is associated with impaired endothelial function later in life [32].
Regarding mechanisms and considering that endothelium-derived nitric oxide regulates arterial elasticity [33], smoking-induced endothelial dysfunction may serve as one of the substrates for the increased aortic stiffness. Cigarette smoking is related to increased endothelin-1, angiotensin II, and thromboxane resulting in vasoconstriction and endothelial dysfunction [34]. Smokers have reduced flow-mediated dilatation [35], whereas sildenafil, a specific type 5 phosphodiesterase inhibitor with vasodilatory effects, abolishes the decrease in FMD of the brachial artery induced by acute smoking [36]. Airflow limitation is associated with higher arterial stiffness. Interestingly, brachial-ankle PWV was an independent determinant of airflow limitation in current and past smokers but not in never smokers with airflow limitation, putting forward an etiological role of smoking in this association [37].
The effect of smoking cessation on arterial stiffness and wave reflections has also been investigated. Arterial stiffness is continuously decreasing along with the duration of smoking cessation. Values of carotid-femoral PWV, AIx, and transit time equal those of nonsmokers after a decade of smoking cessation [29] (Fig. 13.2). Nevertheless, the effect of smoking cessation is more rapidly evident on wave reflections than aortic stiffness. In intervention studies, cessation of smoking for 4 weeks resulted in a decrease in AIx [18], while 6 months of smoking cessation was associated with clear improvement of AIx and corrected heart rate, aortic pressure, and aortic PWV, and with a reduction of daytime BP [20]. In a study using nicotine replacement therapy, improvement of AIx was evident at a short time frame, while at the same time course, no improvement of PWV was demonstrated [38]. Patients who achieved smoking cessation with varenicline (a partial agonist of a4–b2 nicotine acetylcholine receptors) treatment showed significant improvement in central hemodynamics and brachial-ankle PWV, whereas these parameters showed no significant change in those who failed to quit smoking under the same treatment, thus highlighting the beneficial effect of smoking cessation per se on vascular function [39]. Electronic cigarettes are being advocated as tools for smoking cessation with rapidly increasing use. These cigarettes are battery-powered devices that have cartridges or refillable tanks containing a liquid mixture composed primarily of propylene glycol and/or glycerol and nicotine, as well as flavorings and other chemicals. No firm evidence exists on the effect of such an intervention on aortic stiffness and total cardiovascular health, thus no definite recommendations can be made [40].
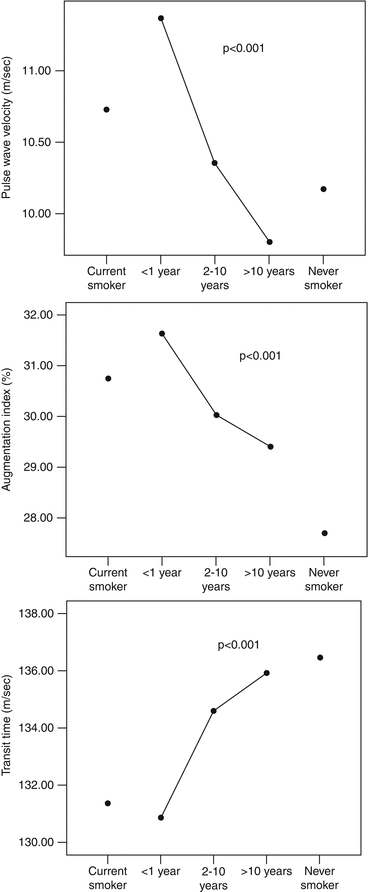
Fig. 13.2
Pulse wave velocity, augmentation index, and transit time of current smokers, never smokers, and ex-smokers (Reproduced with permission from Jatoi et al. [19])
13.2 Diet and Nutrition
13.2.1 Coffee and Caffeine
Coffee is a complex mixture of compounds that may have variable effects on the cardiovascular system. However, the effects of coffee are often confused with the effects of caffeine. Caffeine is a methylated xanthine derivative consumed in coffee, tea, and soft drinks, and it is the most widely used pharmacologically active substance. It should be noted that coffee and caffeine are not interchangeable terms, and results of studies should be interpreted in the context of the particular beverage/substance investigated. Data on the association of coffee consumption with cardiovascular risk are controversial; they are equivocally varying from a positive to a neutral or to even a J- or a U-shaped association [41]. In a recent large study, based upon self-reported drinking habits, two or more cups of coffee per day seemed to reduce all-cause mortality in women (but not in men), primarily due to a reduction in cardiovascular mortality [41]. Similar findings were noted with decaffeinated coffee, suggesting a beneficial effect from a component other than caffeine. The latter appears to be not innocent. Interestingly, regular caffeine intake increases BP, but when ingested through coffee, the BP effect of caffeine is small [42]. The rate of caffeine metabolism through genetic predisposition may be an important determinant of the effect of coffee intake on coronary heart disease risk and account for these discrepancies in outcomes of studies.
Many studies indicate that caffeine intake, in the form of a tablet or in coffee, exerts an acute detrimental effect on aortic stiffness and indices of wave reflections, both in normal subjects and in hypertensive subjects [15, 43–47]. The ingestion of caffeine results in a sustained impairment of arterial stiffness, which lasts longer in hypertensive subjects (at least 3 h) compared with normotensives. Interestingly, the unfavorable effect of caffeine is more evident in central hemodynamics than in peripheral pressure [48] (Fig. 13.3), and coffee increases arterial stiffness to a lesser degree than its contained caffeine [49].
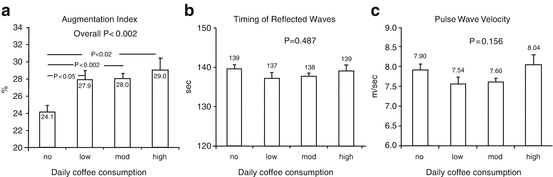
Fig. 13.3
Augmentation index, timing of reflected waves, and pulse wave velocity in different groups of daily coffee consumption (low, <200 ml/day; moderate, 200–450 ml/day; and high, >450 ml/day (Reproduced with permission from Vlachopoulos et al. [48])
Chronic coffee consumption is associated with increased aortic stiffness and wave reflections in healthy normotensives, whereas in hypertensive patients, wave reflections, but not aortic stiffness, are increased [50]. The latter implies that the stiffening effect of coffee in the aorta is probably less prominent in the already stiff aortas of hypertensive patients than in normal aortas.
The caffeine-induced effect on arterial stiffness is probably related to vasoconstriction due to an increase in sympathetic activity and circulating catecholamine concentrations mediated by central nervous system stimulation and antagonism of endogenous adenosine.
Coffee consumption is very frequently combined with smoking. Interestingly enough, the combination of acute caffeine intake/chronic coffee consumption and smoking has a synergistic detrimental effect on carotid-femoral PWV and aortic AIx [15] (Fig. 13.4). Clinical evidence shows that coffee drinking is unrelated to coronary artery disease risk in never smokers, but in ex-smokers and current baseline smokers, daily coffee intake is associated with higher coronary artery disease risk [51].
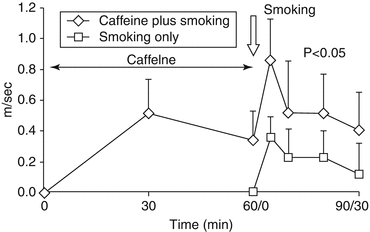
Fig. 13.4
Acute pulse wave velocity responses to caffeine, caffeine plus smoking, or smoking alone. Note the synergistic effect when caffeine and smoking are combined (Reproduced with permission from Vlachopoulos et al. [15])
13.2.2 Salt
therefore, if large amounts of salt are taken,
the pulse will stiffen and harden.
Huang Ti Nei Ching Su Wein, Chinese physician
(1700 BC)
Salt intake is associated with increased BP, and studies have shown a relationship between high salt consumption and increased arterial stiffness. Considering the adverse prognostic role of arterial stiffness, the latter might serve as a potential mechanism, apart from the BP per se, through which salt might be associated with increased cardiovascular risk.
In a seminal study of two Chinese populations, the age-associated increase in carotid-femoral PWV was blunted in the population with a lower salt intake [52]. In insulin-dependent diabetic patients, high sodium intake was associated with decreased femoral arterial distensibility compared to nondiabetic subjects [53]; interestingly, a plausible concept suggests that BP in insulin-dependent diabetes is sodium sensitive and a high sodium intake may be a factor that predisposes to the development of diabetic vascular disease. Although an adverse effect of high salt intake on arterial stiffness is evident in most of the studies, the effect on wave reflections is less clear. Central hemodynamics including central pulse pressure, augmented aortic pressure, and AIx have been independently associated with estimated 24-h sodium excretion, whereas an inverse association was demonstrated between sodium excretion and pulse pressure amplification [54]. However, contrary to these observations, a recent study showed that increase in urinary sodium concentration was independently but inversely associated with central AIx; to further blur the picture, no relationship was established between carotid-femoral PWV and urinary sodium concentrations. Nevertheless, these findings require additional investigation concerning possible mechanistic changes in renal microcirculation after high sodium intake that may explain a possible disassociation between salt intake, aortic stiffness, and wave reflections [55].
Regarding intervention, beneficial effects of low-sodium diet on arterial stiffness have been demonstrated. In older adults with systolic hypertension, sodium restriction led to a rapid increase in large artery compliance, while the accompanying lowering of systolic BP suggests that this improvement in elastic properties may be a key mechanism for the rapid normalization of systolic BP by sodium restriction in such patients [56] (Fig. 13.5). Furthermore, a modest reduction in salt intake for 6 weeks reduced carotid-femoral PWV in mild hypertensives [57]. However, subgroup analysis indicated that the effect was significant only in blacks despite the fact that these subjects achieved a smaller reduction in salt intake and a similar fall in BP compared to whites and Asians, a finding suggesting that the favorable effects of low-sodium diet are more pronounced in salt-sensitive patients. In addition, DASH diet is associated with decreased carotid-femoral PWV [58].
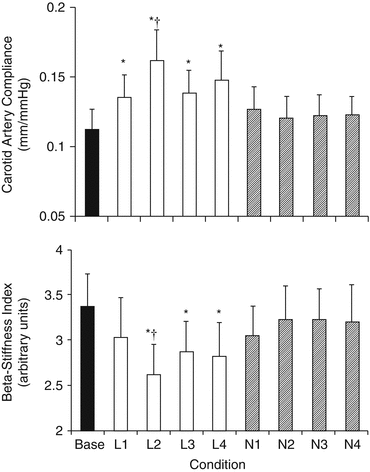
Fig. 13.5
Carotid artery compliance and β-stiffness index at baseline (base), in each of the 4 weeks of the low-sodium condition (L1–L4) and during the 4 weeks of the normal-sodium condition (N1–N4). * Lower than baseline (P < 0.05). † Lower than any given normal week (P < 0.05). The improvement in compliance occurred rapidly, starting by the end of the first week of reduced sodium intake and reaching peak values after only 2 weeks of sodium restriction (Reproduced with permission from Gates et al. [56])
The effect of salt intake on the large arteries may be exerted either through BP-dependent or BP-independent mechanisms. Sodium chloride increases BP, an effect that is due to either an increase in blood volume or an increase in vascular resistance. According to the first observations by Guyton et al. [59], the kidneys have a pivotal role in the pathogenesis of hypertension since the inability of the kidneys to excrete the excess sodium and promote natriuresis is a prerequisite for the BP increase. Sodium retention and the consequent volume expansion initially increase cardiac output. As a result of a process of local autoregulation, peripheral vascular resistance progressively rises so that the long-term sodium-induced BP increase is maintained by an increase in peripheral resistance rather than in cardiac output. Moreover, sodium chloride may also induce alterations in small arteries through endothelial dysfunction as data have shown in resistance arteries of spontaneously hypertensive rats [60]. Furthermore, it also decreases NO synthase activity in rats [61] and enhances the production of endothelin-1 in salt-sensitive hypertensive patients [62]. Additionally, by increasing sympathetic activity, high salt intake may increase the muscular tone of small arteries [63].
A BP-independent relationship of salt intake with arterial stiffness is also likely. An experimental study in anesthetized Dahl rats which were fed either a low- or high-sodium diet for 5 weeks showed that the pressure-compliance curve of the carotid artery was shifted to the right compared with that of Dahl salt-resistant rats or even WKY rats [64]. This finding was evident regardless of the transmural BP. Interestingly, Benetos et al. have also shown that low-sodium diet was associated with an increase in brachial artery diameter after 9 weeks in 20 hypertensive ambulatory patients, a result that was not related to BP fall [65]. On the contrary, no change in carotid artery diameter was observed, thus suggesting a different response of elastic- and muscular-type arteries to salt restriction.
Disassociation of sodium intake with BP changes and a prominent role of the genetic background are emerging. Indeed, data have shown that salt-sensitive hypertensive patients have increased arterial stiffness compared to salt-resistant individuals with the same BP levels, thus indicating that genetic predisposition to salt intake is a strong determinant of the salt-induced vascular changes [66]. In the previously mentioned study of Benetos et al. [64], results also suggest that sodium sensitivity, more than sodium intake, influenced the changes in the carotid artery mechanical properties. Other investigators have shown that in stroke-prone spontaneously hypertensive (SHR-SP) rats, carotid arterial thickness and collagen content were increased and BP was lower compared to SHR-SP rats fed with 1 % saline drinking water, further supporting the notion that genetic sensitivity to sodium, rather than BP itself, influences the changes in arterial structure [67].
Whatever the underlying mechanisms are, the association of arterial stiffness with salt intake is strong. However, although reduction in salt intake reduces arterial stiffness, there might be a threshold of salt consumption under which no further improvement of arterial stiffness is observed. Indeed, in a recent randomized trial in 29 overweight and obese normotensive individuals, no significant change in carotid-femoral PWV was demonstrated when salt intake was reduced from 9.2 to 3.8 g/day for 2 weeks, in spite of a significant improvement in endothelial function [68]. Such a finding raises skepticism about the appropriate recommended daily dose of salt intake. Indeed, associations may be rather complex. The relationship of sodium intake and BP is not linear with age. In a recent study including more than 100,000 adults from 18 countries, sodium intake and BP association was more pronounced not only in patients with hypertension but also in older subjects compared to younger ones [69]. Uncertainty also exists regarding the threshold for CV risk increase. There is evidence of a “J-shaped” relationship of sodium with cardiovascular outcomes. Individuals with low salt intake (less than 2.5 g/day), as well as those with high salt intake (more than 6.0 g/day), shared the same increased cardiovascular risk [70]. However, a more recent study showed that a high burden of deaths from cardiovascular causes was attributed to sodium consumption above a reference level of 2.0 g/day [71]. Until prospective randomized studies provide answers as to whether low or moderate salt consumption has the optimal cardiovascular benefit, the ESH guidelines argue in favor of salt restriction to 5–6 g/day, whereas AHA recommends reducing dietary sodium intake to lower than 1.5 g/day (1 g salt = 400 mg of sodium) [72].
References
1.
Vlachopoulos C, Aznaouridis K, Stefanadis C (2010) Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol 55:1318–1327CrossRefPubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


