PHARMACOLOGY
CHAPTER
Pharmacokinetic and Pharmacodynamic Essentials
Basic understanding of pharmacokinetic and pharmacodynamic concepts is essential to the design of rational, patient-specific pharmacotherapy. The study of pharmacokinetics was first introduced some 40 years ago and is defined as the time course of drug absorption, distribution, metabolism, and elimination. In basic terms, this is described as how the body handles the drug. The concepts of pharmacokinetics can be applied to individual patients in order to maximize efficacy and limit drug toxicities. Drug plasma concentration can help to predict efficacy and toxicity of selected medications. Even though there are limited data for therapeutic drug monitoring for all medications, pharmacokinetic principles can be applied to a wide range of medications.
The primary principle of pharmacodynamics is that a relationship exists between drug concentration at the site of action (receptor) and pharmacologic response. The concentration at the receptor site is most important for elucidating pharmacologic response, with the assumption that serum concentration is directly proportional to receptor concentration. This assumption is not always true. For example, although abciximab has an initial half-life of 10 minutes and second-phase half-life of about 30 minutes, measurement of plasma concentrations of abciximab is not of clinical importance, whereas measurement of platelet activity potentially could be. The focus of this chapter is to review pharmacokinetic and pharmacodynamic concepts and relate them to specific cardiovascular medications. This chapter does not cover current guidelines for use of medications. Please refer to individual chapters in this review book for guideline reference.
PHARMACOKINETICS
Pharmacokinetics refers to the concepts of drug absorption, distribution, metabolism, and elimination (known as “ADME”). Such principles can be applied to drug therapy by such examples as determining loading and maintenance doses of medications, adjusting doses for altered elimination (e.g., renal or hepatic insufficiency), and interpreting plasma drug concentrations. The concepts of ADME are reviewed below.
Absorption
Absorption of medications can occur via multiple routes of administration. Medications administered via the intravenous route are considered to have 100% absorption because the drug is delivered directly into the patient’s circulation. Other routes of administration include oral, transdermal, buccal, sublingual, subcutaneous, intradermal, and rectal. Depending on the type of medication, there are advantages and disadvantages of each of these administration techniques related to absorption. For example, the administration of nitroglycerin via the oral route would provide little systemic effect because of the high degree of presystemic clearance through hepatic metabolism. However, when nitroglycerin is administered intravenously, transdermally, or sublingually, the amount delivered is greatly increased, as presystemic clearance is bypassed.
A number of factors affect the amount of drug absorbed. These include:
 Dose administered
Dose administered
 Fraction of the administered dose that is “active drug” (S)
Fraction of the administered dose that is “active drug” (S)
 Bioavailability of the drug (F)
Bioavailability of the drug (F)
The equation for amount of drug absorbed is
Amount of drug absorbed = (S) (F) (Dose)
The fraction of administered dose that is “active drug” (S) is typically described as the salt form of a drug and varies with different salts. For example, quinidine sulfate has 82% active drug, and quinidine gluconate has 62%. By using the above equation, you can convert one salt form to another salt, assuming you know the bioavailability (F). For example, quinidine sulfate is 82% quinidine base with a bioavailability of 0.73, and quinidine gluconate is 62% quinidine base with a bioavailability of 0.7. With this information, you can compare the amount of quinidine in each tablet to make your conversion. Below is a comparison of quinidine bases, assuming you have 200-mg tablets of both quinidine sulfate (a) andquinidine gluconate (b).
a. Amount of quinidine base absorbed = 0.82 × 0.7 × 200 → 114.8 mg
b. Amount of quinidine base absorbed = 0.62 × 0.7 × 200 → 86.8 mg
Bioavailability (F) is defined as the fraction of an administered dose that reaches the systemic circulation of a patient. Values of bioavailability can be found in a number of pharmacology texts and drug reference handbooks. Factors that can alter bioavailability include:
 Inherent characteristics of the dosage form administered (e.g., tablet dissolution characteristics)
Inherent characteristics of the dosage form administered (e.g., tablet dissolution characteristics)
 Administration route (e.g., oral versus transdermal versus intravenous). The bioavailability of most parenterally administered drugs is 100% (i.e., F = 1).
Administration route (e.g., oral versus transdermal versus intravenous). The bioavailability of most parenterally administered drugs is 100% (i.e., F = 1).
 Issues related to the gastrointestinal (GI) tract
Issues related to the gastrointestinal (GI) tract
The bioavailability of orally administered medications can be affected by gastric pH, GI transit time, gut metabolism, and the presence of food. Certain medications may be unstable in low-pH environments and therefore may be enteric coated in order to prevent breakdown in the stomach. Conversely, certain medications, such as itraconazole, require an acid environment for optimal absorption. Likewise, changes in GI motility with promotility agents or patients experiencing diarrhea may have decreased absorption secondary to decreased transit time through the GI tract. Enzymatic metabolism of medications in the GI tract can also alter absorption and can be responsible for drug interactions. A well-described example of this is the fact that administration of grapefruit juice with certain medications may actually enhance bioavailability secondary to preventing GI metabolism of the compound, thereby increasing the amount of drug available for absorption. Finally, the amount of bioavailable drug may be reduced as a result of the extent of metabolism before reaching the systemic circulation. Examples of this include metabolism via GI bacteria (e.g., digoxin) or “first-pass metabolism” by the liver.
Drugs are absorbed from the GI tract into the portal circulation, and certain drugs are extensively metabolized in the liver before reaching systemic circulation. These drugs have a high first-pass effect or high first-pass metabolism, which can significantly decrease the amount of medication reaching the systemic circulation and hence drug bioavailability. Drugs with high first-pass metabolism have much lower intravenous doses compared to oral doses. Examples of medications with high first-pass metabolism are diltiazem, nitroglycerin, propranolol, verapamil, hydralazine, isoproterenol, labetalol, lidocaine, metoprolol, and nifedipine.
Distribution
After absorption, medications distribute to various tissues in the body to produce a pharmacologic effect. Not all distribution sites for a given medication produce a therapeutic effect. In fact, some distribution sites may produce no effect or untoward effects. The volume of distribution (Vd), or apparent volume of distribution, refers to the total amount of drug in the body, assuming that the drug is present at the same plasma drug concentration (Cp) throughout the body. The Vd is expressed in terms of volume (e.g., liters or liters per kilogram) and is a function of the solubility (lipid versus water solubility) and binding (tissue versus plasma protein binding) characteristics of the drug. Actual sites of distribution cannot be determined from the Vd value.
The volume of distribution equation is
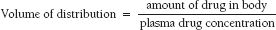
Factors that tend to increase Cp will decrease apparent Vd and include drugs that have:
 Low lipid solubility
Low lipid solubility
 High plasma protein binding
High plasma protein binding
 Low tissue binding
Low tissue binding



