55 Individuals vary widely in their responses to therapy with most drugs. Indeed, response to cardiovascular drug therapy and antiarrhythmics in particular is so highly variable that study of the underlying mechanisms has elucidated important lessons for understanding variable responses to drug therapy in general.1,2 Two key steps are included in the series of events that take place between administration of a drug and manifestation of its beneficial or adverse effects (Figure 55-1). First, drug must be delivered to its molecular site of action (e.g., receptor, ion channel). The magnitude of the effect at the target is determined by drug concentration, and the study of the time dependence of the concentration of drug (and metabolites) achieved in plasma, tissue, or other sites such as urine or bile is termed pharmacokinetics. The second major process that determines drug action has been termed pharmacodynamics and broadly includes the processes that must occur between the interaction of a drug with a specific molecular target and the manifestation of drug action at the molecular, cellular, whole-organ, and whole-patient levels. Because drugs act in a complex (and often abnormal) biological milieu, considerable intersubject variability in drug effects can arise from pharmacodynamic mechanisms. Figure 55-1 Mechanisms modulating drug actions These principles of pharmacokinetics and pharmacodynamics have been recognized for decades, and it is now apparent that they are manifestations of the highly regulated function of individual molecules. Thus, metabolism of a drug occurs by interaction of the drug with specific drug-metabolizing molecules, and absorption, distribution, and renal and biliary excretion reflect cellular drug uptake and efflux by specific transporter3 molecules. It is variability in function and expression of metabolizing and transport molecules, regulated by a host of genetic and environmental factors, that determines pharmacokinetic variability. Similarly, variability in the biological milieu in which drugs act can be conceptualized as variability in the function of multiple molecules, including the target molecules with which drugs interact to produce their beneficial and adverse effects, whose integrated behavior determines normal and abnormal cellular and whole-organ function. Some DNA variants are rare, cause specific “monogenic” diseases, and have conventionally been termed mutations. More common variants are termed polymorphisms and might or might not alter function or expression of the encoded protein. As we begin to understand that each human harbors thousands of DNA variants4,5—some common and some extremely rare—the distinction between “mutation” and “rare variant” becomes increasingly unclear, and more generic language like rare and common polymorphisms is being adopted. One critical aspect of modern genomics is that DNA tends to be highly ancestry specific. Variants implicated in traits like variable drug responses in one ethnic group may be absent in another, or different variants in the same gene may contribute. Associating genetic variants with clinical phenotypes, including drug response, in humans has taken one of two broad approaches. The first is predicated on a perceived understanding of the fundamental physiology, pathophysiology, or pharmacology of the phenotype under study; this is termed a candidate gene approach (see Figure 55-1). The second takes advantage of emerging high-throughput technologies by genotyping or by direct sequencing of large regions of DNA (up to whole exomes and genomes) to then determine whether there is an association between any locus interrogated and the phenotype under study. To date, the most widely used method in this unbiased or hypothesis-free approach is the genome-wide association study (GWAS) paradigm.6 One clear emerging lesson of these genetic association studies is that any result requires further validation both by replication and by further experiments testing the underlying biology. Although the candidate gene approach is intuitively very appealing, repeated experience over the past decade has demonstrated that initially identified associations frequently failed to replicate.7 The reasons for this failure of replication are multiple: (1) The candidate variant may not, in fact, explain a large proportion of the variance in the phenotype under study; (2) the studies generally involve small numbers and so are underpowered; and (3) a publication bias is associated with positive results, so attempts to replicate generally regress to the mean. A major exception to the general “rule” that candidate gene studies fail to replicate in a robust fashion is seen in pharmacogenomics.8 Here, single variants that alter the function of drug-metabolizing or transport molecules may confer a very high likelihood of developing aberrantly high (or low) plasma drug concentrations—and thus highly variable drug responses—during treatment. In addition, genetically determined variations in drug targets (molecules with which drugs interact to achieve their therapeutic or adverse effects) may strongly modulate the outcomes of drug therapy. Specific examples are discussed in the following sections. In arrhythmia science, GWASs have been used to identify new genes and pathways involved in physiological traits (like electrocardiographic [ECG] intervals) and susceptibility to common arrhythmias like atrial fibrillation (AF) or sudden cardiac death.9,10 These results, in turn, are being used to explore the role of variants in these genes in variable drug response. In addition, GWASs have been used to directly analyze drug responses, as described later.11 The GWAS experiment starts by identifying cases and controls for a specific phenotype. These can be categorical (e.g., premature heart disease, breast cancer, drug-induced adverse effect, AF, restless leg syndrome) or continuous (e.g., PR duration, warfarin steady state dose).6 High-throughput platforms are then used to determine genotypes at hundreds of thousands or millions of SNP sites in cases and controls, and tests of association are performed at each SNP to identify those associated with the phenotype under study (Figure 55-2). Evidence that the experiment has yielded a positive result may include very low P values (after correction for multiple comparisons), replication, and ultimately biological plausibility. SNPs are chosen because they tag blocks of linkage disequilibrium; therefore, there is little expectation that those associated with low P values are functional themselves. Rather, they act as sign posts within the genome, identifying specific loci at which functional variants may reside. Figure 55-2 The genome-wide association study paradigm GWAS analyses of the distribution of normal ECG intervals (e.g., PR, QRS, QT) have been conducted in tens of thousands of patients and have identified genomic loci that contribute to variability in these traits.12–18 Some of these are, in retrospect, obvious from an understanding of underlying physiology. Thus, for example, strong signals are present in the KCNQ1 and KCNH2 loci (encoding potassium channels important for cardiac repolarization) in GWAS analyses of variability in the QT interval. Mutations in these genes are the most common causes of the congenital long QT syndrome; the GWAS result demonstrates that common variants in these genes contribute to physiological variability of QT intervals in a normal population. Other signals identified by GWAS identify genes whose role in the phenotype under study is completely unsuspected. In the QT analyses, the strongest signal has consistently been noted near NOS1AP, which encodes an ancillary protein for the neuronal isoform of nitric oxide synthase. Initial studies suggest that NOS1AP encodes a regulator of cardiac potassium and calcium function.19 Follow-up studies have now implicated NOS1AP variants in phenotypes beyond normal QT variability: These include risk for sudden cardiac death in populations,20 risk for events in patients with congenital long QT syndrome,21,22 and risk of sudden death during treatment with some drugs.23 The strongest GWAS signal for variability in PR and QRS is seen in SCN10A, which encodes a sodium channel previously implicated only in pain perception and not known to play a role in the heart. Preliminary studies have suggested multiple roles for the gene in the heart: A contribution to late sodium current,24 regulation of the canonical cardiac sodium channel SCN5A,25,26 and a role in neural regulation of conduction27 have been suggested. GWASs of patients with and without AF have consistently implicated SNPs at chromosome 4q25.28–30 The nearest gene encodes the transcription factor PITX2; initial studies suggest that PITX2c, a cardiac-specific isoform, regulates both development of the pulmonary myocardium31 and expression of other genes (e.g., NPPA, KCNQ1) that have been implicated in AF susceptibility.32 These data are also being used to inform additional studies of variable response to AF therapy. Thus, for example, reports have suggested that SNPs at Chr4q25 predict response to drug33 or ablation34 therapy in AF. In addition to analysis of phenotypes such as ECG intervals or disease susceptibility, the GWAS paradigm has been used to directly study variability in drug response. Here, the problem is that precise definitions of drug response phenotypes are needed, and the numbers of patients that can be accrued is by nature of the experiment much smaller than analyses of ECG intervals or of arrhythmias themselves.11 Nevertheless, as is described further later, initial attempts have been made to analyze phenotypes such as warfarin steady state dose requirement or susceptibility to drug-induced torsades de pointes.
Pharmacogenomics of Cardiac Arrhythmias
Principles of Pharmacogenomics
Definitions
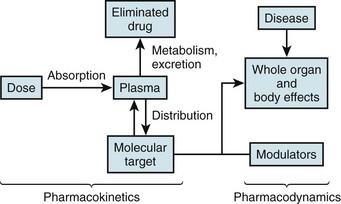
The left side illustrates pharmacokinetic variables that determine absorption, distribution, metabolism, and elimination (often abbreviated ADME). Variability in interactions between drugs and their molecular targets, along with variability in underlying pathophysiologies including disease processes, modulates pharmacodynamic mechanisms (right) that contribute to net drug responses. Understanding the molecular determinants of pharmacokinetics and pharmacodynamics is the first step toward identifying genetic variants that contribute to drug responses.
Approaches to Identifying Pharmacogenetic Variants
Candidate Gene Approaches
Unbiased Approaches: The Genome-Wide Association Study Paradigm
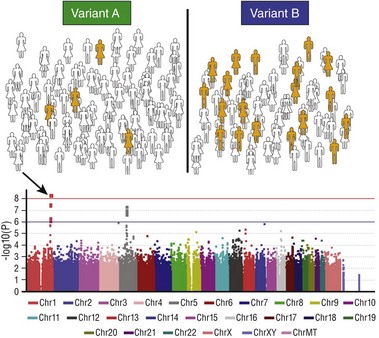
The first step (top panel) is to assign each subject in a large cohort to case (orange) or control (white) status. The entire cohort is then genotyped at hundreds of thousands to millions of common polymorphic sites. The figure illustrates how a hypothetical polymorphism predicting the phenotype in question might segregate: In this case, variant B is associated with the phenotype. A statistical test of association is then performed for each polymorphism and the results displayed on a Manhattan plot (bottom): The x-axis is the chromosomal location of the polymorphism, and the y-axis is the exponent of the P value for the individual statistical test (higher values denote lower P values). In the example shown, a cluster of polymorphisms in chromosome 1 (arrow) achieves P values less than 10−8. The red horizontal line denotes an arbitrary level of statistical significance after correction for multiple testing.
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pharmacogenomics of Cardiac Arrhythmias
